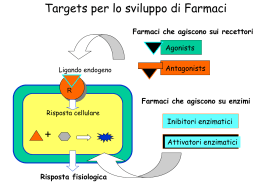
UNIVERSITA' DEGLI STUDI DI PISA FACOLTA' DI SCIENZE MATEMATICHE FISICHE E NATURALI DIPARTIMENTO DI NEUROSCIENZE CORSO DI LAUREA IN SCIENZE BIOLOGICHE L’ATTIVITÀ DI AGONISTA PARZIALE DEGLI ANTIPSICOTICI ARIPIPRAZOLO, BIFEPRUNOX, NDESMETILCLOZAPINA, S33592 E PRECLAMOLO PER IL RECETTORE DOPAMINERGICO D2L È MODIFICATA DALLA COTRANSFEZIONE CON IL RECETTORE D3: RUOLO POTENZIALE DELLA FORMAZIONE DI ETERODIMERI RELATORE Prof. Roberto Maggio CANDIDATA Maria Laura Grieco ANNO ACCADEMICO 2005/2006 1 ABSTRACT By analogy to preclamol, the novel antipsychotics, aripiprazole and bifeprunox, the major metabolite of clozapine, Ndesmethylclozapine, and the benzopyrollidine derivative, S33592, all behave as partial agonists at D2 and, though less wellcharacterised, D3 receptors. However their actions at D3/D2 heterodimers have not, to date, been examined, an issue addressed in COS7 cells expressing recombinant, dopamine D2L and/or D3 receptors. Employing [3H]nemonapride, all ligands showed comparable affinity for D3 and D2L sites, except aripiprazole which revealed 10 fold preference for the latter. In D2L receptorexpressing cells cotransfected with a D3 receptor insensitive, chimeric adenylyl cyclaseV/VI, all drugs reduced forskolinstimulated cAMP production by 1525% as compared to quinpirole (48%). Further, quinpiroleinduced inhibition was attenuated by aripiprazole and S33592 to a level corresponding to their effects alone. In cells cotransfected with D3 and D2L receptors, quinpirole displayed similar potency and efficacy as in D2Ltransfected cells, whereas aripiprazole, S33592, bifeprunox, Ndesmethylclozapine and preclamol were inactive: furthermore, aripiprazole and S33592 abolished the actions of quinpirole. Corroborative data were acquired with D2 trunk/D3 tail and D3 trunk/D2 tail chimeras where the agonist actions of quinpirole were similarly blocked by aripiprazole and S33592 which were, like bifeprunox, Ndesmethylclozapine and preclamol, inactive alone. By contrast, when a 12 amino acid sequence in the third intracellular loop of D3 receptors was replaced by the corresponding sequence of D2L receptors, aripiprazole, S33592, bifeprunox, N desmethylclozapine and preclamol behaved as partial agonists (ca 20%) as compared to quinpirole (42%). Moreover, at D2L receptorexpressing cells cotransfected with modified D3i3(D2) receptors, all drugs partially inhibited ACV/VI activity compared to quinpirole. To summarize, aripiprazole, S33592, bifeprunox, Ndesmethylclozapine and preclamol behave as partial 2 agonists at D2L receptors coupled to AC: their antagonist actions upon coexpression of D3 with D2L receptors probably reflects physical association (heterodimer formation) and weakened coupling efficacy. These findings have interesting implications for the functional and clinical profiles of these antipsychotic agents. 3 1. INTRODUZIONE 1.1 Dopamina La dopamina è il neurotrasmettitore che nel sistema nervoso centrale (SNC) dei mammiferi ha un ruolo importante in numerose funzioni tra cui l’attività motoria, l’apprendimento, l’emozioni, la gratificazione, la motivazione e la secrezione di alcuni ormoni come la prolattina. Inoltre, possiamo ipotizzare anche una funzione endogena della dopamina nel sistema nervoso periferico (SNP) per la presenza di recettori dopaminergici a livello della muscolatura liscia vascolare a livello renale, a livello del sistema gastroenterico ed infine a livello delle fibre gangliari del sistema simpatico. L’importanza del sistema dopaminergico centrale nel controllo dell’attività motoria è chiaramente dimostrata nella malattia di Parkinson dove la degenerazione di neuroni dopaminergici presenti nell’area A9 del mesencefalo (nigra pars compacta) è la causa principale della malattia. Alterazioni a carico del sistema dopaminergico sono anche ipotizzate in disturbi psichiatrici di tipo psicotico, come la schizofrenia, dove l’uso di antagonisti dei recettori D2 dopaminergici risulta essere uno dei trattamenti farmacologici più efficaci. L’uso invece di antagonisti dopaminergici poco liposolubili, che non passano la barriera ematoencefalica, nel trattamento di disturbi di tipo gastroenterico o come farmaci antiemetici dimostra l’importanza del sistema dopaminergico anche a livello periferico. Negli ultimi anni è stato chiaramente indicato come il sistema dopaminergico è anche coinvolto nei meccanismi neurofisiologici che sono alla base delle tossicodipendenze. I precursori della sintesi di dopamina nel neurone dopaminergico sono gli aminoacidi tirosina e fenilalanina che sono assunti con le proteine presenti nella dieta. Questi due aminoacidi attraversano la barriera ematoencefalica grazie ad un trasportatore di membrana per gli Laminoacidi aromatici e successivamente sono captati all’interno del neurone dopaminergico attraverso lo stesso trasportatore. All’interno del neurone, l’aminoacido fenilalanina è trasformato in tirosina e successivamente quest’ultima è convertita dall’enzima tirosina idrossilasi in L3,4 diidrossifenilalanina (LDopa). La tirosina idrossilasi è un marker istologico specifico dei neuroni catecolaminergici e la sua attività è finemente regolata, diventando così l’enzima limitante la sintesi delle catecolamine. 4 La dopamina sintetizzata exnovo, è immagazzinata nelle vescicole sinaptiche mediante un trasportatore vescicolare e da qui rilasciata nel vallo sinaptico per esocitosi Ca++dipendente in seguito all’arrivo di stimoli eccitatori che depolarizzano il neurone (Fig. 1). Fig . 1. Trasmissione dopaminergica 5 Infine, dopo aver svolto la sua azione legandosi ai recettori specifici, essa è captata dal neurone dopaminergico presinaptico dove può essere nuovamente immagazzinata nelle vescicole o può essere catabolizzata per opera di due isoforme enzimatiche delle monoaminoossidasi (MAO)A e MAOB. Una via metabolica alternativa di degradazione della dopamina è quella operata dall’enzima catecolOmetiltransferasi (COMT) all’esterno del neurone presinaptico (Fig. 2). Queste due vie metaboliche operano congiuntamente e convergono portando alla formazione dell’acido omovanillico (HVA), il metabolita principale del catabolismo della dopamina (Vedi Testo Farmacologia generale e molecolare, Clementi; UTET, 1999). La dopamina inoltre può subire una trasformazione non enzimatica per ossidazione e dar luogo a composti potenzialmente neurotossici. Gli eventi cellulari e molecolari coinvolti nella tossicità della dopamina all’interno dei neuroni dopaminergici potrebbero dare origine a fenomeni degenerativi che determinano morte cellulare (Corsini et al., 2002). Gli effetti di questa catecolamina a livello del sistema nervoso centrale sono mediati dalla sua interazione con i recettori dopaminergici. Questi fanno parte della grande famiglia di proteine di membrana rappresentata dai recettori accoppiati a proteine G, Gproteincoupled receptors (GPCR)s. 6 Fig . 2. Sintesi e metabolismo della dopamina. 1.2 Recettori accoppiati a proteine G : Struttura I recettori accoppiati a proteine G (GPCRs) rappresentano il sistema di traduzione del segnale più diffuso nel regno animale. La maggior parte delle molecole che fungono da messaggeri tra cellula e cellula (ormoni, neurotrasmettitori, neuromodulatori) inducono risposte combinandosi a recettori accoppiati a proteine G. Le diverse famiglie di GPCRs hanno tra loro una struttura molto simile, ciò risulta evidente dall’esame delle sequenze primarie. I GPCRs sono costituiti da una singola catena polipeptidica che attraversa sette volte la membrana con l’estremità amminoterminale extracellulare e quella carbossiterminale intracellulare. Le sette regioni transmembrana ad αelica, unite tra loro da tre loops intracellulari e tre extracellulari, si dispongono a formare un core idrofobico. Tra le diverse famiglie le caratteristiche strutturali che variano sono la composizione e la lunghezza del terzo loop citosolico e dell’estremità carbossiterminale. Il legame con ligandi specifici avviene tramite interazioni multiple tra gruppi funzionali del ligando e aminoacidi altamente conservati presenti nei domini extracellulari e/o nei domini transmembrana del recettore. 7 Fig.3. struttura di un recettore accoppiato a proteina G Esistono tre classi di recettori accoppiati a proteine G che tengono conto delle diversità dovute sia alla sequenza aminoacidica sia alla modalità d’interazione con il ligando (Bockaert et al., 1999). 8 La famiglia 1 raggruppa la maggior parte dei recettori, tra cui la rodopsina, ed è ulteriormente suddivisa in tre sottofamiglie. La famiglia 1a comprende i recettori per i ligandi piccoli, come i muscarinici, i dopaminergici o gli adrenergici ed il sito di legame è localizzato all’interno della tasca recettoriale formata dai sette segmenti transmembranali. La famiglia 1b è rappresentata da recettori per i peptidi, il cui sito di legame è presente nella parte Nterminale, nelle anse extracellulari e nella parte superiore dei segmenti transmembrana; nella classe 1c troviamo i recettori per gli ormoni lipoproteici caratterizzati da un’estremità Nterminale lunga che insieme alla parte esterna del primo e secondo loop extracellulare costituiscono il sito di legame. All’interno della famiglia 2, i recettori accoppiati a proteine G hanno una morfologia simile a quella del gruppo 1c ma non ne condividono la stessa similitudine nella sequenza aminoacidica. I ligandi fisiologici per i recettori di questo gruppo sono ormoni ad alto peso molecolare come il glucagone o la secretina. Infine, la famiglia 3 include i recettori metabotropici al glutammato, i recettori sensibili al Ca++, recettori GABAb e i recettori per alcuni ferormoni. In questa famiglia, la lunga estremità Nterminale è determinante per l’interazione con i ligandi endogeni. Un contributo importante per la comprensione della struttura tridimensionale dei recettori accoppiati alle proteine G è stato dato grazie alla determinazione della struttura cristallina del recettore per la rodopsina (Palczewski et al., 2000; Fig. 4). 9 10 Fig . 4. Struttura tridimensionale del recettore per la rodopsina vista lateralmente (A) e dal basso (B e C). 1.3 Recettori accoppiati a proteine G : traduzione del segnale L’interazione ligandorecettore induce un cambiamento conformazionale della proteina recettoriale, in particolare a livello del secondo loop citoplasmatico (Burstein et al., 1998; Kozell et al., 1994), delle estremità N e Cterminale del terzo loop citoplasmatico e della coda Cterminale citosolica. La traduzione del segnale coinvolge, oltre al recettore, due elementi: una proteina G eterotrimerica (costituita da tre subunità: α, β e γ) che lega i nucleotidi guanosinici GDP e GTP, e un bersaglio proteico che funge da effettore. Il legame con il neurotrasmettitore promuove interazioni allosteriche tra il recettore e la proteina G inducendo lo scambio del nucleotide guanosinico GDP con il GTP sulla subunità α . Questo destabilizza il complesso trimerico αβγ con conseguente dissociazione della subunità α, legata al GTP, dal dimero βγ. La proteina G “attivata” è così in grado di agire sull’effettore (Fig. 5). La fine del segnale avviene tramite idrolisi del GTP in GDP per merito della stessa subunità α . Esistono vari tipi di proteine G (almeno una ventina sono note) suddivise in base al tipo di risposta che inducono, come ad esempio: Gs, attivazione dell’enzima adenilato ciclasi Gi, inibizione dell’adenilato ciclasi e apertura di canali al K+ Go, inibizione di canali al Ca++ voltaggiodipendenti Gq, attivazione della fosfolipasi Cβ Dopo che la proteina G ha interagito con uno dei suddetti effettori, vengono prodotti secondi messaggeri (cAMP, IP3/DAG, acido arachidonico) che sono responsabili della risposta cellulare conseguente allo stimolo recettoriale. La parte Cterminale della subunità α della proteina G sembra essere determinante nel conferire selettività all’interazione col recettore (Conklin et al., 1993; Conklin et al., 1996), sebbene un ruolo ausiliare può essere svolto anche dal complesso βγ (Florio e Sternweiss, 1985; Butkerait et al., 1995). 11 Nonostante l’interazione recettoreproteina G sia piuttosto selettiva, alcuni recettori sono in grado di attivare più di un tipo di proteina G (Ganze t al., 1990; Eason et al., 1992; Eason e Ligget, 1995). Questa promiscuità nell’accoppiamento è dovuta soprattutto al corredo di proteine G che una determinata linea cellulare esprime, poiché il legame tra un recettore con una bassa selettività per una determinata proteina G può essere forzato se in quella cellula è espresso prevalentemente un tipo di proteina G. Per esempio i recettori dopaminergici possono mostrare alcune proprietà tradizionali in certi tipi cellulari che non sono confermate in altre linee, a dimostrazione della complessità e della varietà di interazione coinvolte tra recettore e proteine G. 12 Fig . 5. Eventi intracellulari attivati dalle proteine G. La specificità del recettore verso le varie proteine G è determinata soprattutto dalle parti N e Cterminale del terzo loop citoplasmatico (Wess et al., 1997). Questi dati sono stati confermati da esperimenti condotti su chimere recettoriali D1/D2 (Kozell et al., 1994) e D2/D3 (Lachowicz et al., 1997; Filteau et al., 1999). Inoltre, un ruolo importante per l’accoppiamento tra recettore e proteina G è stato dimostrato anche per il secondo loop intracellulare (Kozell et al., 1994). Recenti studi hanno dimostrato come, attraverso la loro capacità di regolare effettori specifici, i recettori accoppiati alle proteine G possono indurre eventi intracellulari simili a quelli attivati dai recettori per i fattori di crescita ad attività tirosina chinasica (Vedi Testo Clementi; UTET, 1999). Una delle più importanti conseguenze di questa stimolazione è l’attivazione di alcune chinasi specifiche chiamata MAP chinasi (Mitogenactivated protein kinases), che svolgono un ruolo fondamentale nel controllo della proliferazione e del differenziamento cellulare. I recettori accoppiati alle proteine G sono in grado di attivare le MAP chinasi con meccanismi molteplici tuttora oggetto di studio. Una prima possibilità, utilizzata soprattutto dai recettori accoppiati alle proteine Gi, sembra coinvolgere il complesso βγ ed agisce sulle proteine adattatrici Shc. Un’altra possibilità è quella di indurre una fosforilazione nei residui tirosinici dei recettori per i fattori di crescita con un meccanismo ancora da caratterizzare definito “transattivazione”. I recettori accoppiati a proteine Gi possono stimolare le MAP chinasi grazie anche all’azione sulla proteina chinasi C, che è in grado di fosforilare la proteina Raf, attivando la cascata di fosforilazioni intracellulari senza interagire con la proteina di membrana Ras (Fig.6). 13 Fig . 6. Attivazione di MAP chinasi indotta dai GPCRs. 14 1.4 Recettori dopaminergici La prima evidenza sperimentale, che ha dimostrato l’esistenza dei recettori per la dopamina nel sistema nervoso centrale (SNC), risale a studi biochimici compiuti nel 1972 che rilevarono la capacità del neurotrasmettitore di stimolare l’enzima adenilato ciclasi (Kebabian et al., 1979) In seguito, sulla base di risultati farmacologici, i recettori dopaminergici sono stati classificati in due classi dette D 1 e D2 (Spano et al., 1978). L’avvento delle nuove tecnologie di DNA ricombinante ha consentito di riconoscere l’esistenza di diversi sottotipi recettoriali dopaminergici che, in base sia alle omologie di sequenza che al profilo funzionale e farmacologico, possono ancora essere suddivisi nelle due classiche famiglie D1 e D2. Appartengono alla famiglia D1 (D1like) i recettori D1e D5 e alla famiglia D2 (D2like) i recettori D2, D3 e D4. I recettori D1like sono sensibili alla dopamina, apomorfina e ad altri agonisti come il fenoldopam e bloccati da antagonisti specifici come lo SCH23390. I recettori dopaminergici D2 e D3 sono stimolati da vecchi e nuovi farmaci antiparkinsoniani come bromocriptina, pergolite, apomorfina, pramipexolo e cabergolina, anche se è possibile rilevare una certa preferenza d’affinità verso i recettori D3. Una buona selettività nei confronti del recettore D3 è mostrata anche da alcuni composti di sintesi come il quinpirolo e il 7OHDPAT. Per quanto riguarda gli antagonisti recettoriali, i neurolettici sulpiride e aloperidolo presentano una preferenza verso il sottotipo recettoriale D2, mentre i recettori D4 sono molto sensibili alla clozapina (Fig. 7) . La difficoltà nel differenziare farmacologicamente i vari sottotipi recettoriali D2like è dovuta soprattutto all’elevata omologia di sequenza aminoacidica. I recettori D1 e D5 mostrano una elevata omologia nelle sequenze di aminoacidi nei domini transmembrana (80%), regioni deputate al legame dei farmaci. Per quanto riguarda le regioni transmembrana dei D2like, i recettori D2 e D3 mostrano una omologia di sequenza per il 72%, mentre i D2 e D4 per il 53%. L’estremità aminoterminale consiste di un numero simile di aminoacidi in tutti i sottotipi recettoriali e presenta una quantità variabile di siti di glicosilazione. La coda carbossiterminale è circa sette volte più lunga nei recettori D1like rispetto ai D2like ed è ricca di residui di serina e treonina. La difficoltà nel differenziare farmacologicamente i vari sottotipi recettoriali D2like è dovuta soprattutto all’elevata omologia di sequenza aminoacidica. 15 Al contrario, i recettori D2like hanno il terzo loop intracellulare più lungo, caratteristico dei recettori che interagiscono con la proteina Gi, mentre quello dei D1like è più corto come in molti recettori che legano la proteina Gs. SISTEMA FAMIGLIA GENICA SOTTOTIPO AGONISTI ANTAGONISTI apomorfina SCH39930 DI TRASDUZIONE D1 D1 ↑adenilato ciclasi fenoldopam pergolide D5 D2 (long e short) bromocriptina come D1 come D1 lisuride come D1 ↓adenilato ciclasi apomorfina aloperidolo ↑canali K+ pergolide sulpiride ↓canali Ca++ pramipexolo raclopride come D2 D2 D3 come D2 quinpirolo come D2 7OH DPAT D4 come D2 come D2 come D2 clozapina Fig . 7. Classificazione farmacologica dei recettori dopaminergici nel SNC 16 1.4.1 I Recettori D1like Sottotipo D1 Il recettore D1 è una glicoproteina con due siti di glicosilazione, uno nella parte Nterminale e l’altro nel secondo loop extracellulare. Presenta inoltre un sito di fosforilazione cAMPdipendente nel terzo loop citoplasmatico e un residuo di cisteina nella estremità carbossiterminale (altamente conservato nei GPCRs) che viene complessato con l’acido palmitico. Questo legame con l’acido palmitico, che àncora la coda Cterminale del recettore alla membrana, forma il quarto loop intracellulare. Ulteriori siti di fosforilazione possono essere i residui di serina e treonina che si trovano all’estremità Cterminale. Questo sottotipo recettoriale è quello maggiormente espresso tra i diversi recettori dopaminergici a livello del sistema nervoso centrale sia di ratto che di uomo (Missale et al., 1998). La presenza di mRNA del recettore D1 è stata determinata nel nucleo caudato e nel putamen (nuclei del sistema extrapiramidale), nel nucleo accumbens e nel tubercolo olfattorio (che fanno parte del sistema limbico) ed inoltre è stato identificato in minor quantità nella corteccia cerebrale, nel talamo e nell’ipotalamo. Da un punto di vista funzionale il recettore D1 è accoppiato alla proteina Gs, per cui attiva l’adenilato ciclasi. Il recettore D1 sembra essere in grado di modulare i livelli di calcio intracellulare attraverso differenti meccanismi. In alcuni sistemi cellulari il meccanismo di azione coinvolge la produzione di IP3 grazie alla attivazione della fosfolipasi C, mentre in altri sistemi l’effetto sulla concentrazione di Ca++ intracellulare sembra passare da un meccanismo diverso che consiste nell’attivazione della PKA attraverso l’aumento di cAMP. Aumenti dell’attività di alcune MAP chinasi per azione del recettore D 1 sono stati riportati su p38 e JNK in alcune linee cellulari ma non su ERK12 (Zhen et al., 1998), anche se l’effetto del cAMP nelle cellule COS sull’attivazione di ERK12 lascia pensare che il recettore D1 potrebbe attivare anche ERK12 (Faure et al., 1994). Un effetto del recettore D1 sull’attivazione di ERK12 è stato recentemente riportata in vivo su un modello di parkinsonismo animale, dove la carenza di dopamina nello striato induce una particolare sensibilità ad agonisti D1 selettivi (Gerfen, 2002). Sottotipo D5 17 Il recettore D5 è stato isolato e clonato da Sunahara et al. (1991). È omologo per il 50% al recettore D1 e nelle sequenze transmembrana l’omologia aumenta fino all’80%. Vari agonisti e antagonisti hanno simile affinità per i due recettori ad eccezione della dopamina che è più affine di circa dieci volte verso il recettore D5 rispetto a quello D1. Questo recettore è localizzato per lo più nell’ippocampo e nell’ipotalamo, ma in generale i suoi livelli di espressione sono molto inferiori rispetto a quelli del sottotipo D1. Le caratteristiche funzionali di questo recettore sono simili a quelle del recettore D 5, soprattutto per quanto riguarda la formazione di cAMP. 18 1.4.2 I Recettori D2like Sottotipo D2 Il recettore D2 presenta i sette domini transmembrana tipici dei GPCRs, all’estremità Nterminale ha tre siti di glicosilazione, all’estremità Cterminale ha una cisteina per il legame con l’acido palmitico e nel terzo loop intracellulare ha un sito di fosforilazione per una chinasi cAMPdipendente. Inoltre sul terzo loop citoplasmatico, ci sono altri siti di fosforilazione importanti nella regolazione del turnover recettoriale (Missale et al., 1998). Esso è presente in due diverse varianti chiamate D2long e D2short che sono originate da uno splicing alternativo di un esone di 87 bp localizzato tra gli introni 4 e 5. Le proteine D2long e D2short si differenziano rispettivamente per la presenza o meno di una sequenza di 29 aminoacidi nel terzo loop intracellulare. Per queste due isoforme non è ancora stata identificata una differenza farmacologica, ma solo una lieve diversità funzionale. Esse mostrano un simile pattern di distribuzione, ma la forma D2short è meno abbondantemente rappresentata nei tessuti. Il recettore D2 è maggiormente localizzato nei nuclei caudato e putamen (quindi a livello dello striato), nel nucleo accumbens e nel tubercolo olfattorio. Nel 50% circa dei neuroni di media grandezza del caudato, il D2 è coespresso con il D1. Inoltre, è stato dimostrato che la colocalizzazione D1/D2 porta ad un’azione sinergica dei due recettori (Pomelli et al., 1991). Il D2 è presente anche nelle cellule lattotrope ipofisarie, dove media l’inibizione dopaminergica della secrezione di prolattina. Da un punto di vista funzionale il recettore D2 è accoppiato alla proteina Gi e quindi inibisce la formazione di AMP ciclico. In diversi tipi cellulari, come i neuroni mesencefalici o in cellule provenienti dall’ipofisi anteriore, il recettore dopaminergico D2 fa aumentare l’uscita dello ione potassio con conseguente iperpolarizzazione della cellula, attraverso l’attivazione di proteine G sensibili alla tossina della pertosse. Il significato funzionale della iperpolarizzazione mediata dai recettori D2 in questi neuroni sembra essere l’inibizione del release di dopamina. Questi recettori in questi neuroni funzionano da autorecettore dopaminergici. Sono stati riportati anche effetti del recettore sulle correnti al Ca ++, soprattutto di tipo inibitorio, e sulla cascata dell’acido arachidonico. Il recettore D2 è in grado di potenziare il rilascio di acido arachidonico indotto dall’aumento di Ca++ intracellulare (Missale et al., 1998). 19 Numerose evidenze sperimentali hanno dimostrato come i recettori D2like, in particolare il recettore D2, siano coinvolti anche nella mitogenesi e nel differenziamento cellulare. Inoltre, il recettore D 2 è in grado di stimolare l’attività di ERK12 in differenti linee cellulari come le CHO, COS e C6. Se il recettore D2 viene espresso in cellule mesencefaliche “immortalizzate”, l’effetto di dopaminoagonisti è quello di influenzare la vitalità neuronale intesa come numero di neuriti (Swarzenski et al.,1993), probabilmente grazie all’attivazione delle MAP chinasi. Tuttavia, in uno studio dove sono state utilizzate colture primarie di neuroni mesencefalici, gli agonisti dopaminergici non hanno avuto effetto sulla sopravvivenza o sul differenziamento di queste cellule (Van Muiswinkel et al., 1993). Nel modello di emiparkinsonismo di ratto trattato con 6OHDA, l’effetto della rotazione controlaterale indotta dall’agonista D2 selettivo è bloccata dall’inibizione di MAP chinasi dimostrando un ruolo importante di tale proteina nell’effetto comportamentale dovuto alla somministrazione di farmaci antiparkinsoniani (Cai et al., 2000). Sottotipo D3 Il recettore D3 è stato inizialmente clonato da una libreria di cDNA di ratto usando sonde derivate dalla sequenza del recettore dopaminergico D2 (Sokoloff et al.,1990). È infatti un recettore molto simile al D2 sia per quando riguarda la sequenza aminoacidica che per l’organizzazione strutturale nella membrana.. Anche per il recettore D3 sono state identificate varianti di splicing. Nel topo esistono due varianti con uno splicing alternativo di 21 amminoacidi a livello del terzo loop citoplasmatico (Fishburn et al., 1993) mentre nel ratto e nell’uomo esistono altre varianti, forse non funzionali, di cui parleremo in seguito (Seeman e Van Tol, 1994). Il recettore D3 di ratto è presente soprattutto nello striato ventrale (tubercolo olfattorio, nucleo accumbens, isole di Calleja) e nell’ipotalamo a dimostrazione di una sua importanza nelle funzioni limbiche dell’animale. Esso si trova in quantità minore anche in altre aree cerebrali come la substantia nigra, lo striato dorsale e la corteccia prefrontale. Queste indicazioni sono state date misurando la quantità di mRNA con la tecnica del Northern Blot (Sokoloff et al., 1990) e utilizzando un composto selettivo marcato come il [3H]7OHDPAT a basse dosi su un tessuto cerebrale (Levesque et al., 1992). Se la misurazione di un mRNA del recettore D3 è piuttosto specifica, le indicazioni di binding con il [3H]7OHDPAT lasciano adito a qualche dubbio poiché potrebbero essere influenzate dall’interazione del ligando radiomarcato con il recettore D2 (Landwehrmeyer et al., 1992). Nel ratto e nel topo la quantità del recettore D3 rispetto al D2 misurato con la tecnica del binding è notevolmente inferiore. Per esempio se consideriamo lo striato dorsale la differenza è di circa cento volte, mentre nello striato ventrale la differenza si riduce a venti volte. Queste misurazioni non escludono comunque la possibilità che in alcuni sottogruppi neuronali le differenze di espressione tra D3 e D2 siano minori, come è stato indicato per esempio nello shell del nucleo accumbens dell’ animale. 20 Da notare che l’espressione del recettore D3 aumenta in modo rilevante nello striato dorsale denervato per trattamento con LDopa (Borden et al., 1997 e 2000). Tale aumento evidenziato in un modello di parkinsonismo sperimentale di ratti trattati con 6OHDA, è stato invocato come possibile causa del fenomeno della sensibilizzazione alla LDopa, comportamento paragonabile alle discinesie nei primati. Se passiamo dalla specie dei roditori a quella dei primati, come l’uomo per esempio, assistiamo ad un drastico aumento della quantità di recettore D3, tanto che è stato stimato che nell’uomo la sua espressione possa essere inferiore rispetto al D2 di solamente 35 volte circa (Suzuki et al., 1998; Joyce et al., 1999). Inoltre nell’uomo misurando la quantità di mRNA, è stato osservato come la distribuzione del recettore D 3 sia diffusa anche in regioni corticali, nel nucleo caudato e putamen, nel talamo, nell’ippocampo e nella substantia nigra pars compacta (Fig. 8). 21 22 Fig . 8. Espressione di mRNA del recettore D3 nell’uomo Da un punto di vista funzionale il recettore D3 condivide in parte le caratteristiche del recettore D2 e pertanto inibisce la formazione di AMP ciclico attraverso l’attivazione della proteina G i. Tuttavia in molti sistemi cellulari, il recettore D3 è incapace di inibire l’adenilato ciclasi (Tang et al., 1994; Freedman et al., 1994) e quindi mostra, in generale, d’accoppiarsi più debolmente alla proteina Gi rispetto al recettore D2 (Vanhauwe et al.,1999). Un fattore determinante per la funzionalità del recettore D3 è probabilmente il tipo di adenilato ciclasi con la quale interagisce. Nelle cellule COS, per esempio, la presenza dell’adenilato ciclasi V permette al recettore D3 di funzionare correttamente, mentre esso risulta debolmente accoppiato all’adenilato ciclasi di tipo II (Caron et al., 1997). Questo recettore dopaminergico sembra anche modulare le correnti uscenti di ione potassio, come dimostrato in colture cellulari mesencefaliche (Tang et al., 1994). Tale effetto iperpolarizzante è probabilmente responsabile dell’effetto inibitorio del recettore sul release di dopamina nelle cellule mesencefaliche. Il recettore D3, come gli altri recettori D2like, è coinvolto probabilmente nella mitogenesi e nel differenziamento, come dimostrato in alcune linee cellulari osservando l’incorporazione di [ 3H]timidina (Griffon et al., 1997). Il meccanismo attraverso cui il recettore D3 induce l’aumento dell’attività di sintesi del DNA potrebbe essere quello della fosforilazione delle MAP chinasi, attraverso la stimolazione di una isoforma di protein chinasi C (Cussan et al., 1999). Sottotipo D4 L’ultimo recettore ad essere stato clonato è il recettore D 4 (Gelernter al., 1991). Questo ha una struttura molto simile agli altri due recettori D2like. È localizzato principalmente nella corteccia frontale, nel mesencefalo e nell’amigdala, ma si trova in tracce anche nello striato e nel tubercolo olfattorio. I suoi livelli di espressione nel sistema nervoso centrale comunque, così come per il sottotipo D3, sono in misura minore rispetto al D2. Una caratteristica di tale recettore nell’uomo è la presenza di un polimorfismo genetico a livello del terzo loop citosolico che consiste nella ripetizione di 16 aminoacidi (Van Tol et al.,1992). Tra le varie isoforme genetiche non sono state riscontrate differenze funzionali. Le caratteristiche funzionali sono paragonabili al recettore D2. 23 1.5 Dimerizzazione dei recettori accoppiati a proteine G Negli ultimi anni un numero considerevole di lavori ha dimostrato l’esistenza di recettori accoppiati alle proteine G in forma omodimerica ed eterodimerica (Agnati et al., 2005; Bouvier et al., 2001). Numerose evidenze sperimentali di tipo biofisico e biochimico hanno confermato la presenza di numerosi complessi recettoriali in cellule “viventi” supportando l’idea che la dimerizzazione possa essere un processo importante per la biogenesi e la funzione dei recettori accoppiati alle proteine G. Se da una parte l’omodimerizzazione apre nuove questioni sui meccanismi molecolari coinvolti nella trasduzione del segnale evocato dal neurotrasmettitore, la presenza di eterodimeri potrebbe portare alla formazione di nuove strutture recettoriali con profili funzionale e farmacologici nuovi, contribuendo in tal modo all’interazione tra diversi sistemi neurotrasmettitoriali. Tenendo conto che i recettori accoppiati a proteine G sono il maggior “target” nel campo farmaceutico, l’esistenza di dimeri potrebbe avere delle implicazioni per lo sviluppo di nuovi farmaci. D’altronde il concetto di dimerizzazione come fenomeno coinvolto nell’attivazione di recettori transmembrana è stato dimostrato per altre proteine recettoriali e costituisce spesso una tappa decisiva per un’appropriata risposta biologica. Un esempio importante è rappresentato dai recettori tirosinchinasi per i fattori di crescita (EGFR, PDGFR e FGFR) che trovano nella dimerizzazione il proprio meccanismo di attivazione. Per quanto riguarda i recettori accoppiati a proteine G, essi sembrano essere particolarmente versatili e dinamici all’interno della propria struttura proteica e ciò potrebbe consentire una più facile interazione recettorerecettore. Diversi esperimenti hanno dimostrato che i GPCRs sono complessi proteici costituiti da più subunità indipendenti che interagiscono tra loro. È possibile infatti frammentare questi recettori in due parti in grado di riconoscersi e ricongiungersi sulla membrana cellulare per ricostituire il recettore originale. Questo fenomeno è stato dimostrato per diversi recettori, come per esempio il recettore per la rodopsina (Ridge et al., 1995), quello β2adrenergico (Kobilka et al., 1998), i recettori muscarinici M2 e M3 (Maggio et al., 1993), il recettore per la vasopressina V2 (Shoneberg et al., 1995), quello per l’ormone del rilascio delle gonadotropine (Grosse et al., 1997), quelli alle neurochinine NK1 (Nielsen et al., 1998) e il recettore D2 per la dopamina (Scarselli et al., 2000). Il taglio effettuato a livello del terzo loop intracellulare dà luogo a due distinti frammenti recettoriali: a) il frammento “trunk”, contenente l’estremità Nterminale, i domini transmembrana da I a V e l’estremità Nterminale del terzo loop 24 citoplasmatico; b) il frammento tail cpontenente il Cterminale del terzo loop citoplasmatico le regioni transmembrana VI e VII e l’estremità Cterminale del recettore. I frammenti recettoriali ottenuti sono capaci di riconoscersi e riassociarsi fra di loro ricostituendo il recettore wildtype dal quale derivano. Come descritto sotto, questo meccanismo di riconoscimento ed interazione tra i due frammenti recettoriali potrebbe essere alla base di uno dei meccanismi di interazione recettorerecettore. Questo modello di dimerizzazione recettoriale proposto è il cosiddetto “domain swapping” (scambio di domini), in cui la parte trunk di uno dei due monomeri interagisce con quella tail dell’altro. Il risultato è un dimero recettoriale con una struttura proteica tetramerica, a quattro subunità, stabilizzata da un numero d’interazioni chimiche praticamente doppio rispetto a quello presente nel recettore monometrico. Questo tipo di interazione è stato proposto per spiegare la dimerizzazione tra due recettori chimerici inattivi formati da parte del recettore muscarinico M3 e parte dal recettore adrenergico α2 (Maggio et al., 1993). Tale lavoro è particolarmente importante perché ha dimostrato per la prima volta la possibilità di un’interazione diretta a livello molecolare tra due recettori accoppiati alle proteine G. In particolare, la chimera M3/α2 era formata da i primi V segmenti transmembrana del recettore M3 e dai segmenti VI e VII del recettore α2, mentre la chimera α2/M3 risultava costruita in modo complementare (Fig. 9). Le due chimere recettoriali erano incapaci di legare i vari composti adrenergici e muscarinici ed inoltre erano inattive funzionalmente. La loro coespressione invece portava al recupero funzionale sia del recettore M3 che di quello α2, dimostrando quindi che le due chimere inattive interagivano tra loro ripristinando la funzione dei recettori wild type. Questo meccanismo di interazione è suffragato da evidenze sperimentali e da modelli computazionali (Gouldson et al., 1998). 25 26 Fig. 9. Meccanismo del “domain swapping” (da: Protein eng. 1998, Vol. 11) 27 1.5.1 Evidenze di dimeri recettoriali Il primo esempio di eterodimero recettoriale fisiologico evidenziato è stato il recettore GABAb. In questo caso sono state individuate e clonate due differenti sequenze geniche del recettore GABAb nel cervello di ratto, GABAbR1 e GABAbR2, dimostrando che per ottenere una piena attività recettoriale le due subunità erano entrambe fondamentali (Jones et al., 1998). La presenza di una sola delle due subunità nella cellula non era sufficiente per ottenere la funzione del recettore GABAb, mentre la coespressione delle due consentiva di ottenere un recettore completamente funzionante. Questo fu il primo esperimento che dimostrava come l’interazione tra le due subunità di un recettore fosse un requisito indispensabile per il corretto funzionamento della trasduzione del segnale di un neurotrasmettitore a livello sinaptico. È stato anche dimostrato, utilizzando la microscopia elettronica, come le due subunità recettoriali GABAbR1 e GABAbR2 fossero colocalizzate in vivo nel ratto a livello delle spine neuronali delle cellule di Purkinje del cervelletto (Kaupmann et al., 1998), suggerendo come l’interazione tra i due recettori possa veramente essere un fenomeno fisiologico estendibile nel regno animale. Studi successivi hanno confermato la necessità della presenza di entrambe le subunità per un corretto funzionamento del recettore GABAb (Mitrovic et al., 2001; Gelvez et al., 2001; Havlikova et al., 2002), indicando anche gli aspetti molecolari di questa interazione. In particolare è stato dimostrato come la presenza di entrambe le subunità sia fondamentale per il corretto “traffincking” recettoriale ed inoltre sono state individuate le funzioni dei sottotipi recettoriali. Se la subunità GABAbR1 è fondamentale per l’iterazione con i ligandi gabaergici, la subunità GABAbR2 è essenziale per l’accoppiamento con le proteine G e per il raggiungimento dell’espressione su membrana. Tuttavia, questa netta distinzione di ruoli delle due subunità non è stata ancora del tutto dimostrata dato che alcuni studi hanno evidenziato come nella struttura eterodimerica anche il recettore GABAbR1 contribuisca all’effetto funzionale mentre il recettore GABAbR2 aumenti l’affinità verso alcuni agonisti gabaergici. Oggi gli studi eseguiti sulla presenza dell’eterodimero GABAbR1/GABAbR2 sono quelli maggiormente convincenti, confermati e difficilmente confutabili, che incoraggiano fortemente a proseguire su questo filone di ricerca con la speranza di scoprire nuovi eterodimeri recettoriali. 28 Se lo studio sul complesso eterodimerico gabaergico ha aperto la strada nel campo della dimerizzazione dei GPCRs, gli esperimenti sui recettori oppiodi μ, δ e κ, hanno indicato come questo fenomeno molecolare di interazione fra recettori possa dare origine alla formazione di complessi proteici con caratteristiche funzionali e farmacologiche distinte dai recettori che li formano (Devi et al., 2000). L’interazione molecolare diretta di tipo eterodimerico tra i recettori δ e κ porta alla formazione di un complesso proteico con caratteristiche funzionali e farmacologiche distinte. Inoltre, l’eterodimero δ/κ è capace di legare sinergicamente due ligandi oppiodi selettivi potenziando il segnale di trasduzione della fosforilazione di MAP chinasi (Jordan et al., 1999). La farmacologia di questo sito eterodimerico è differente da quella dei recettori δ e κ che lo compongono, mentre risulta molto vicina a quella del sottotipo recettoriale precedentemente caratterizzato come recettore κ 2. tali indicazioni sperimentali danno conferma all’ipotesi che la formazione di eterodimeri potrebbe davvero, in alcuni casi, dare origine a nuovi siti recettoriali con caratteristiche farmacologiche proprie. Esperimenti analoghi sono stati fatti sui recettori μ e δ, ottenendo risultati simili. La farmacologia del sito eterodimerico μ/δ sembra molto vicina a quella del sottotipo recettoriale δ 2 (Gomes et al., 2000). Dal punto di vista funzionale sono state rilevate nuove caratteristiche del sito eterodimerico, addirittura in alcuni casi opposte rispetto ai recettori μ e δ che lo compongono (Charles et al., 2003). La scoperta di recettori eterodimerici oppiodi ha portato ad un notevole interesse per eventuali applicazioni cliniche. Per esempio è stato dimostrato che somministrando in vivo contemporaneamente agonisti selettivi per i recettori μ e δ si ottiene un effetto sinergico antidolorifico dovuto ad un potenziamento della funzione recettoriale. La morfina, un potente antidolorifico, è un agonista μ e può indurre dipendenza e tolleranza; se viene somministrato congiuntamente un agonista δ, quest’ultimo potrebbe potenziare l’effetto analgesico della morfina consentendo una diminuzione della sua dose. Una possibile spiegazione dell’effetto sinergico ottenuto, somministrando contemporaneamente un μ agonista e un δ agonista, è che entrambi siano in grado di attivare congiuntamente il recettore eterodimerico μ/δ. La formazione di un nuovo sito recettoriale è stata evidenziata anche peri recettori muscarinici M2 e M3. la coespressione dei due recettori muscarinici porta alla formazione di un sito eterodimero con caratteristiche farmacologiche simili a quelle di un recettore chimerico formato dalla parte trunk del recettore M3 e dalla parte tail del recettore M2, indicando come meccanismo possibile quello del “domain swapping” (Maggio et al., 1999). Finora abbiamo trattato dell’interazione tra sottotipi recettoriali che fanno parte dello stesso sistema neurotrasmettitoriale, tuttavia esempi d’interazione tra recettori che rispondono a differenti neurotrasmettitore non mancano. Nel caso particolare è stato dimostrato come i recettori dopaminergici D2 e i recettori alla somatostatina SSTR5 interagiscano fisicamente attraverso l’eterodimerizzazione creando un nuovo recettore ad alta affinità capace di legare 29 contemporaneamente somatostatina e agonisti dopaminergici (Rocheville et al., 2000). L’aumento di affinità di tale complesso eterodimerico è circa 30 volte e l’interazione tra i due recettori è stata determinata in cellule viventi utilizzando la tecnica della FRET. Dati di questo tipo sono estremamente interessanti, perché potrebbero contribuire alla spiegazione del fenomeno della cotrasmissione. Cotrasmettitori come la somatostatina o altri possono influenzare l’attività di sistemi neurotrasmettitoriali classici, come quello della dopamina per esempio, il meccanismo d’interazione potrebbe almeno in parte passare dalla formazione di eterodimeri recettoriali in grado di captare la presenza di due ligandi diversi. l’eterodimerizzazione di recettori accoppiati a proteine G, oltre ad influenzare le funzioni e le affinità dei recettori, potrebbe svolgere un ruolo importante nei fenomeni di internalizzazione e desensitizzazione dei recettori sulla membrana cellulare. Se vengono coespressi, per esempio, nella stessa cellula recettori oppiodi δ e recettori adrenergici, osserviamo che l’agonista adrenergico internalizza entrambi i recettori (Jordan et al., 2001). La presenza dell’eterodimero δ/β2 sensibile ad entrambi gli agonisti potrebbe spiegare questo fenomeno di cooperazione. D’altra parte il fenomeno sopra descritto risulta specifico considerando che in cellule che esprimono sia il recettore κ che il recettore β2 non si verifica tale interazione. Indicazioni della stessa natura sono state suggerite per i recettori eterodimerici SSTR2a/μ (Pfeiffer et al., 2002). In questo caso la stimolazione con somatostatina o con un ligando oppiode del recettore eterodimerico non solo influenza l’internalizzazione di entrambi i recettori, ma anche il grado di fosforilazione e desensitizzazione di entrambi. Quindi l’eterodimerizzazione tra recettori accoppiati a proteine G si presenta come un fenomeno biologico funzionale importante almeno nei sistemi cellulari sperimentali; rimane da confermare tale indicazione negli organismi animali viventi come l’uomo per esempio. Dati sperimentali sull’uomo del fenomeno della eterodimerizzazione in realtà sono stati ottenuti, ma non come meccanismo di una funzione fisiologica quanto come possibile causa di patogenesi. Questo non significa che l’eterodimerizzazione in vivo abbia solamente un significato fisiopatologico, ma dimostra come tale meccanismo biologico sia estremamente diversificato a seconda del sistema recettoriale di cui ci stiamo occupando. È stato dimostrato come nelle gestanti che soffrono di una patologia come la preeclampsia si osserva una risposta esagerata agli effetti pressori dell’angiotensina II e tale effetto potrebbe passare dalla presenza dell’eterodimero formato tra il recettore all’angiotensina II AT1 e il recettore alla bradichinina B2 (Abdalla et al., 2001). l’eterodimerizzazione tra AT1 e B2 è correlabile con un aumento di circa cinque volte la quantità del recettore B2; inoltre, l’espressione dell’eterodimero AT1/B2 fa aumentare la risposta all’angiotensina II e conferisce resistenza al recettore AT1 nei normali processi di disattivazione indotta delle specie reattive dell’ossigeno. 30 Se finora sono state analizzate interazioni tra recettori accoppiati alle proteine G, uno studio ha evidenziato come sia possibile un’interazione diretta anche tra recettori accoppiati alle proteine G e recettori canale. Tale interazione è stata dimostrata tra il recettore dopaminergico D5 e il recettore canale GABAA (Liu et al., 2000). La formazione dell’eterodimero D5/GABAA potrebbe spiegare in parte alcune interazioni funzionali tra il sistema neurotrasmettitoriale dopaminergico e quello gabaergico. Da questi esempi di eterodimerizzazione, dal significato così diverso, emerge come questo meccanismo biologico debba ancora essere ben caratterizzato e studiato in dettaglio soprattutto in sistemi nativi; tuttavia dalle indicazioni finora ottenute possiamo ipotizzare che il fenomeno possa essere importante e costituisca un ulteriore meccanismo di regolazione e modulazione nel complesso sistema d’interazione tra neurotrasmettitori e recettori. 1.5.2 Meccanismi di dimerizzazione recettoriale Per quanto riguarda la struttura tridimensionale dei dimeri recettoriali, l’unica evidenza diretta proviene da studi cristallografici sui recettori metabotropici al glutammato mGluR1 (Kunishima et al., 2000). In questo studio è stata determinata la struttura tridimensionale cristallina della parte extracellulare Nterminale sia in presenza che in assenza di glutammato. La struttura cristallina ha dimostrato che un ponte di solfuro tra le due cisteine (Cys140) è responsabile della connessione tra i due monomeri, anche se interazioni tra vari segmenti proteici ad αelica contribuiscono alla stabilità del dimero. Il complesso omodimerico mostra una certa flessibilità e dinamicità conformazionale ed il ligando stabilizza la struttura attiva del recettore. L’omodimerizzazione di alcuni recettori metabotropici al glutammato e di recettori Ca++sensibili avviene quindi con un meccanismo di formazione di un ponte disolfuro, ma probabilmente sono coinvolte anche interazioni di tipo idrofobico tra differenti segmenti proteici ad αelica (Fig. 10) Un altro meccanismo di dimerizzazione è stato messo in luce nell’interazine tra due i due sottotipi recettoriali GABAbR1 e GABAbR2. Un’interazione tra le due code recettoriali (Coleidcoil) a livello del Cterminale è stata ipotizzata nella formazione dell’eterodimero grazie a studi di mutagenesi (Margeta et al., 2001). Questi studi hanno anche dimostrato che, sebbene l’interazione “coleidcoil” sia importante per la corretta espressione recettoriale in membrana, essa non è necessaria per la formazione del dimero. 31 Il terzo meccanismo di dimerizzazione tra recettori monomerici è quello che prevede un’interazione soprattutto di tipo idrofobico a livello dei segmenti transmembrana come dimostrato da studi di mutagenesi sul recettore β 2 adrenergico e D2 dopaminergico. Insieme a questi studi, analisi di simulazione computazionale hanno suggerito che siano soprattutto i domini transmembrana V e VI che creano l’interfaccia di interazione tra i due monomeri (Gouldson et al., 1998). In realtà il meccanismo di interazione tra le parti transmembrana dei due recettori potrebbe avvenire con due meccanismi diversi, uno che prota alla formazione di “Contact Dimers” e l’altro di “Swapping Dimers”. Nel meccanismo del “domain swapping” (scambio di domini), domini identici di due recettori vengono a scambiarsi tra un monomero e l’altro. Il risultato è un dimero recettoriale con una struttura proteica tetramerica, formata da quattro ipotetiche subunità che generano due siti recettoriali, stabilizzata da un numero d’interazioni chimiche praticamente doppie rispetto a quello presente nel recettore monometrico. Questo scambio è reso possibile dalla libertà di movimento che le due porzioni hanno all’interno dello stesso monomero recettoriale, dovuta sia alla lunghezza del terzo loop intracellulare, sia alla fluidità del doppio strato fosfolipidico di membrana. 32 Fig. 10. Meccanismo di interazione recettoriale: Domain swapping (A), interazione tra segmenti transmembrana (B), formazione di ponti disolfuro (C) e interazione “coiledcoil” (D). Un esempio di questo tipo di interazione è stato dimostrato tra i recettori muscarinici M 2 e M3. il sito eterodimerico M2/M3 ha mostrto una farmacologia paragonabile a quella di un recettore chimerico muscarinico fatto dalla parte trunk del recettore M2 e la parte tail del recettore M3. Nel caso dei “contact dimers” invece, si suppone un’interazione lateroleterale, in cui i recettori monomerici prendono contatto tra loro attraverso la parte idrofobia delle regioni transmembrana. Un’interazione di questo tipo è stata ipotizzata per l’eterodimero formato dal recettore D2 e quello SSTR5 per la somatostatina. In questo complesso recettoriale, l’agonista dopaminergico determina un potenziamento dell’affinità per la somatostatina di circa trenta volte. 1.6 SISTEMA DOPAMINERGICO: PATOLOGIE E FARMACI I neuroni dopaminergici sono rappresentati nel sistema nervoso centrale da diversi gruppi cellulari distinti in base alla loro funzione e localizzazione: • Il sistema dopaminergico nigrostriatale, che ha i suoi corpi cellulari di origine nel mesencefalo ventrale, comprende: i neuroni A10 dell’area tegmentale ventrale (VTA) mesencefalica, i neuroni A9 della pars compacta della substantia nigra e i neuroni A8 dell’area retrorubrale in posizione più caudale. I neuroni A9 nella pars compacta della substantia nigra rappresentano l’origine della componente dorsale del sistema mesostriatale. I dendriti di questa popolazione neuronale innervano la pars reticolata della substantia nigra dove il rilascio di dopamina regola l’attività delle terminazioni afferenti originate dai gangli della base. Gli assoni dei neuroni A9 proiettano al nucleo caudato e al putamen che insieme costituiscono il corpo striato. • Il sistema dopaminergico mesolimbico e mesocorticale, e la componente ventrale del sistema dopaminergico mesostriatale originano soprattutto nei neuroni A10 della VTA e nella parte mediale della substantia nigra. Questa componente innerva il nucleo accumbens, il tubercolo olfattorio e il nucleo interstiziale della stria terminalis. Altre fibre che originano dai neuroni A10 innervano il setto (soprattutto il 33 nucleo laterale del setto), l’ippocampo, l’amigdala, la corteccia entorinale, la corteccia prefrontale, la corteccia peririnale e la corteccia piriforme. • Il sistema dopaminergico mesotalamico ha i suoi neuroni di origine nell’area A10; questi neuroni innervano le strutture del Fig. 11. Vie dopaminergiche del sistema nervoso centrale di ratto ponte, del diencefalo e del telencefalo. Un fascio mesotalamico molto ben caratterizzato origina nella VTA e termina nell’abenula, in particolare nelle sue parti laterale e mediale. • I sistemi dopaminergici tuberoinfundibolare e tuberoipofisario originano dai corpi cellulari dei neuroni dopaminergici, detti neuroni A12, localizzati nei nuclei arcuato e periarcuato dell’ipotalamo. Il sistema tubero ipofisario origina nella parte anteriore dell’area A12 e innerva la parte intermedia e posteriore dell’ipofisi, dove inibisce rispettivamente la secrezione dell’ormone melanocitostimolante (αMSH) e della βendorfina, e il rilascio degli ormoni ossitocina e vasopressina. I neuroni del sistema tuberoinfundibolare innervano lo strato esterno dell’eminenza mediana, dove sono strettamente in contatto con i capillari del sistema portale ipofisario; la dopamina rilasciata nel sistema portale ipofisario, raggiunge l’ipofisi anteriore in cui media l’inibizione della secrezione di prolattina. A tutti questi livelli, un’alterazione della neurotrasmissione dopaminergica può portare alla genesi di vari disturbi del sistema nervoso centrale. Tra questi, il morbo di Parkinson dovuto alla degenerazione dei neuroni dopaminergici del 34 sistema nigrostriatale; alcune forme di psicosi e la dipendenza psichica verso le sostanze d’abuso, per alterazioni del sistema mesolimbico e mesocorticale. Nella cura del morbo di Parkinson e di alcune psicosi, la terapia farmacologia attualmente disponibile è principalmente mirata su composti affini per i recettori D2like. Nel trattamento del morbo di Parkinson, inoltre, viene utilizzata la L DOPA, precursore della biosintesi della dopamina, capace di reintegrare i livelli del neurotrasmettitore che è andato perduto a causa della degenerazione dei neuroni dopaminergici. In seguito a trattamenti prolungati questo farmaco perde di efficacia e compaiono quelle che vengono chiamate fluttuazioni motorie e discinesie. Sebbene non si conosca con precisione la causa di questa caduta dell’effetto terapeutico del farmaco, l’ipotesi più accreditata è quella che presuppone la drastica riduzione dei neuroni dopaminergici a livello nigrostriatale dopo alcuni anni di trattamento. Questo farebbe sì che la maggior parte della LDOPA non possa venire trasformata in dopamina e accumulata nei neuroni dopaminergici della nigra. Una quota esigua di dopamina potrebbe tuttavia originare da una trasformazione aspecifica della LDOPA all’interno di neuroni non dopaminergici. Per questo motivo, la ricerca farmacologica del morbo di Parkinson è molto attiva nel cercare dei farmaci agonisti dopaminergici ad azione diretta sui recettori, che non dipendano per la loro attività da una trasformazione metabolica e quindi dalla presenza di neuroni dopaminergici integri. Tra gli agonisti che sono stati sintetizzati e che sono entrati in clinica per il trattamento di questa patologia ricordiamo la bromocriptina, la pergolide, l’apomorfina, il ropinirolo e il pramipexolo. Questi farmaci sembrano ritardare l’esordio degli effetti collaterali da trattamento cronico con la LDOPA. Alcune psicosi come la schizofrenia, al contrario, sembrano derivare da un’iperattività dopaminergica del sistema meso limbico, e quindi per la terapia vengono somministrati farmaci con effetto antagonista dopaminergico, conosciuti anche come neurolettici. Essi rappresentano una classe di farmaci eterogenea da un punto di vista chimico, ma hanno tutti la capacità di antagonizzare i recettori dopaminergici di tipo D2 e D3. I neurolettici vengono distinti in classici e atipici. I primi causano molti effetti collaterali per via dell’inattivazione indiscriminata dei recettori dopaminergici striatali e ipofisari con induzione di sindrome parkinsoniana, amenorrea e ginecomastia. Gli atipici invece hanno ridotti effetti collaterali di tipo extrapiramidale e ipofisari, inoltre essi hanno un più ampio spettro di attività recettoriale. Al momento attuale non è ancora ben chiaro quale sia il meccanismo d’azione dei farmaci neurolettici atipici. 35 2. SCOPO DELLA TESI Nella mia tesi ho testato come la eterodimerizzazione dei recettori dopaminergici D2 e D3 modifica l’attività farmacologia di una serie di agonisti parziali per questi due recettori. I composti che sono stati testati sono: l’aripiprazolo, un nuovo farmaco antipsicotico ormai in commercio da alcuni anni, e quattro agonisti parziali usati solo a scopo sperimentale, il preclamolo, il bifeprunox, il più importante metabolita dell’antipsicotico atipico clozapina, l’Ndesmetilclozapina e l’ S33592 (un derivato benzopirrolidinico) sintetizzato dalla casa farmaceutica Servier. L’efficacia di questi agonisti parziali è stata valutata sia sui recettori D2 e D3 transfettati singolarmente che sui due recettori cotransfettati nelle stesse cellule. 36 3. MATERIALI E METODI Il materiale utilizzato per il terreno di coltura cellulare (DMEM, Lglutammina, aminoacidi non essenziali, penicillina e streptomicina, Glucosio, Timidina e Ipoxantina, e Metotrexato ) è stato acquistato dalla Sigma Chemical Company (St.Louis, MO, U.S.A.), il siero fetale bovino dializzato è stato comperato dalla Invitrogen ( Gibco). La [3H]Adenina e la [3H]Nemonapride sono state fornite da DuPontNew England Nuclear (Boston, MA), la forscolina e quinpirolo sono state acquistate dalla Sigma Chemical Company . Aripiprazolo, preclamolo, S33592, bifeprunox ed Ndesmetilclozapina sono state sintetizzate da G. Laville e JL. Peglion (Servire, Paris, France). NHCOH3 N NC N Cl Cl O N H O N O Aripiprazole S 33592 HO N Préclamol 37 H N N N Quinpirole 3.1 COLTURE CELLULARI Sono stati utilizzati tre tipi cellulari, le cellule COS7 per le transfezioni transienti e le cellule CHO per quelle stabili. In particolare sono state usate delle cellule CHO deficienti per il gene dell’enzima diidrofolato reduttasi (DHFR) e cellule CHO provviste del geni per l’enzima e per il recettore dopaminergico umano D3 (DHFR+/HD3). I due tipi cellulari CHO sono stati forniti dal Dott. Pierre Sokoloff ( Sokoloff et al. , 1992 ). Le cellule CHO DHFR sono state ottenute in seguito a mutagenesi con Etil Metasulfonato e con raggi γ e sono state selezionate aggiungendo al medium di coltura Diidrossiuridina triziata e Metotrexato ( Urlaub et al.,1980 ). Le cellule CHO DHFR+ HD3 sono state ottenute trasfettando cellule CHO DHFR con un plasmide contenente la sequenze per l’enzima diidrofolato redattasi e per il recettore HD3. Il recettore HD3 è stato ottenuto da screening di librerie di DNA con sonde costituite da sequenze di recettore D3 di ratto e usando la tecnica della polymerase chain reaction (PCR). IL cDNA così ottenuto è stato inserito nel plasmide vettore pGEM4Z (Promega) a livello del sito di restrizione BglΙΙ ( incluso nel primer usato per la PCR). 38 Il cDNA è stato poi subclonato in un vettore eucariotico derivato dal plasmide pSV contenente la sequenza per la Diidrofolatoreduttasi (Sokoloff et al., 1990) usando gli enzimi di restrizione KpnΙ e BglΙΙ . 39 L’enzima diidrofolato reduttasi (DHFR) è responsabile della sintesi dell’acido tetraidrofolico intracellulare , un cofattore necessario per il trasferimento di unità monocarboniose in un gran numero di reazioni biosintetiche in cui questi gruppi vengono trasferiti da un metabolita ad un altro. Alcune di queste complesse reazioni sono state descritte nel metabolismo intermedio degli amino acidi, delle purine e delle pirimidine. Le cellule sono state fatte crescere in flasche da 150 cm2 e una volta raggiunta la confluenza sono state staccate con tripsina 0,2 % e seminate in piastre petri da 100 mm. Negli esperimenti funzionali dopo la transfezione le cellule sono state riseminate in piastre da 24 pozzetti per l’esecuzione del saggio dell’adenilato ciclasi che di norma veniva eseguito dopo 48 ore dall’ultima semina. Le cellule sono state cresciute in atmosfera umidificata al 5 % di CO2 e ad una temperatura di 37ºC. Per la crescita e la proliferazione delle cellule CHO i mezzi di coltura sono stati adattati come descritto qui di seguito. 40 Il terreno per le CHO DHFR contiene : Dulbecco’s modified Eagles Medium (DMEM ) supplementato con Siero fetale bovino dializzato (FBS), glucosio, 2% di L glutammina 200 mM, 1% di aminoacidi non essenziali (Lalanina, Lasparagina, acido L aspartico, acido Lglutammico, glicina, Lprolina, Lserina), l’1% di una soluzione di penicillina (100 unità/ml) e streptomicina (10 mg/ml). Inoltre a tale terreno è stata addizionata una soluzione di TimidinaIpoxantina . Le DHFR sono incapaci di sintetizzare de novo glicina, nucleotidi purinici e timidilato, per questo il mezzo di coltura è opportunamente modificato in modo da permettere la crescita cellulare. Il terreno di coltura delle CHO DHFR+ è uguale a quello per le CHO DHFR tranne che per la presenza di Metotrexato 150 nM e per l’assenza di Timidina Ipoxantina. Il Metotrexato è un analogo del tetraidrofolato e blocca l’attività della diidrofolatoreduttasi la cui sintesi viene quindi incrementata con un meccanismo a feedback . 41 fig12. Immagine di cellule CHO in coltura 3.2 VETTORE PLASMIDICO Il vettore plasmidico contenente la sequenza codificante per il recettore HD2 è stata fornita dal Guthrie cDNA Resource Center. Il recettore HD2 ( variante 1 ) è stato clonato in pcDNA3.1+ (Invitrogen) , l’inserzione è stata effettuata tra i siti di restrizione EcoRΙ (5') e XhoΙ (3'). Il vettore contiene il gene per la resistenza alla Neomicina, per la selezione di linee cellulari che stabilmente esprimono il plasmide. La selezione è fatta con G418. Il G418 è un antibiotico aminoglucosidico simile alla Neomicina, che blocca la sintesi proteica nelle cellule di mammifero interferendo con la funzione ribosomale. Uno chema del plasmide è riportato nella figura nella pagina seguente. Il plasmide presenta un sito di riconoscimento per la replicazione batterica (pUCori), il promotore, il sito di origine e quello di poliadenilazione del virus SV40. Il promotore che permette l’espressione della proteina ricombinante inserita nel sito policlonale è quello del Citomegalovirus (Pcmv), mentre il sito di poliadenilazione per il distacco dell’enzima RNA polimerasi dal DNA è quello dell’ormone bovino della crescita (BGH pA). 42 43 44 3.3 TRANSFEZIONE TRANSIENTE Per la transfezione transiente sono state usate le cellule COS7. Esse sono state distribuite in piastre Petri di 100 mm di diametro ad una densità di 4 x 10 5 cellule in 10 ml di terreno di coltura. Passate 24 ore dalla semina, le cellule sono state lavate per due volte con una soluzione sterile di NaCl allo 0,9%, dopodiché sono state transfettate transientemente con il metodo del DEAEdestrano (Cullen 1987). In breve, lo NaCl residuo è stato aspirato ed è stata aggiunta una soluzione costituita da: 540 μ l di tampone salino fosfato (PBS) sterile (cloruro di calcio 0.133 g/l, cloruro di magnesio 0.1 g/l, fosfato di potassio monobasico 0.2 g/l, fosfato di potassio dibasico 1.15 g/l, cloruro di sodio 8 g/l, pH 7.4), 28 μ l di una soluzione di DEAEdestrano (10 mg/l) e diverse concentrazioni (0.25 μ g, 0.5 μ g, 1 μ g, 2 μ g, 3 μ g e 4 μ g) di DNA plasmidico. Dopo un periodo di incubazione di 30 minuti a 37°C, la soluzione è stata rimossa e sono stati aggiunti nelle piastre 6 ml di terreno di coltura e 60 μ l di clorochina 8 mM (80 μ M finale). Le piastre sono state nuovamente messe a incubare per 2 ore e mezzo, dopodiché questa soluzione è stata rimossa e le cellule sono state lavate per due volte con una soluzione sterile di NaCl al 0.9%. Alla fine sono stati aggiunti 10 ml di terreno di coltura fresco, e le piastre sono state rimesse nell’incubatore. Per gli esperimenti di binding le piastre sono state utilizzate dopo tre giorni, mentre per gli esperimenti funzionali 24 ore dopo la trasfezione le cellule sono state riseminate in piastre da 24 pozzetti come descritto sopra. 3.4 TRANSFEZIONE STABILE Entrambe le linee cellulari CHO sono state trasfettate stabilmente con il recettore HD2 in modo da ottenere cellule CHO DHFR HD2 e cellule CHO DHFR+ HD3/HD2 . La trasfezione stabile del vettore contenente il gene per il recettore HD2 è stata ottenuta con il metodo del Calcio Fosfato (Jordan et al., 1996 ). Le cellule sono state coltivate in piastre di 100 mm di diametro ad una densità di circa 105 cellule per piastra in 10 ml di mezzo di coltura. Il giorno seguente è stato aspirato il medium e, senza fare lavaggi , è stato sostituito con 9 ml di medium fresco . Un’ora dopo è stata effettuata la trasfezione : per ogni piastra è stata preparata una soluzione contenente 50 μl di CaCl2 2,5 M e 440 μl di TE Buffer (1mM Tris HCl, 0.1mM EDTA pH 7.7). A questa soluzione sono stati poi aggiunti 10 μg di DNA e 500 μl di Hepes 2x (140 mM NaCl , 1.5 mM Na2HPO4 , 50 mM Hepes , pH 7.05). Il tutto è stato lasciato ad 45 incubare per 30 secondi. La soluzione così preparata è stata aggiunta goccia a goccia ai 9 ml di medium già presenti nelle piastre da trasfettare. In seguito alla trasfezione in alcune cellule il DNA plasmidico viene integrato permanentemente nel DNA nucleare. Le cellule che hanno integrato stabilmente il cDNA trasfettato sono state selezionate sfruttando la resistenza alla geneticina presente nel plasmide pCDNA3.1. 46 3.5 METODICA PER GLI STUDI DI BINDING DEI RECETTORI Trascorsi tre giorni dalla transfezione, le piastre sono state lavate per due volte con soluzione fisiologica e trattate con 2 ml di tampone ipotonico freddo (Na+HEPES 1 mM, EDTA 2 mM). Dopo 20 min di incubazione in ghiaccio a 4°C, le cellule sono state staccate e trasferite in opportuni tubi per poi essere centrifugate a 17.000 rpm per 20 min a 4°C. I pellets di membrana sono stati successivamente omogeneizzati con un politron settato a velocita V per 30 sec in un tampone così costituito: TrisHCl 50 mM, pH 7.4, NaCl 155 mM, albumina bovina 0.01 mg/ml. In seguito i pellets sono stati aliquotati per le prove di binding. Gli esperimenti di saturazione sono stati condotti utilizzando la [3H]nemonapride con attività specifica di 82 Ci/mmol. La determinazione del binding aspecifico è avvenuta in presenza di dopamina alla concentrazione di 2 mM. Per quanto riguarda le prove di spiazzamento le membrane sono state risospese nel medesimo tampone addizionato di Tris HCl 2 mM, NaCl 7,70 mM pH7,4 e di acido ascorbico allo 0.025%. Le sostanze testate negli esperimenti di inibizione sono state: Preclamolo, Aripiprazolo, Ndesmetilclozapina, S33592 e Bifeprunox. Tutti gli esperimenti sono stati condotti in un volume finale di 1 ml. L’incubazione è stata effettuata a temperatura ambiente per 1 h, e per separare il radioligando legato al tessuto da quello non legato, i campioni sono stati filtrati con un raccoglitore “Brandel Cell Harvester” utilizzando filtri a fibre di vetro (Whatmann, GF/B). Successivamente i filtri sono stati lavati tre volte con un tampone freddo (TrisHCl 10 mM, pH 7.4, NaCl 155 mM). Infine i filtri sono stati trasferiti in tubi contenenti 4 ml di liquido di scintillazione e la radioattività associata al recettore è stata valutata mediante il conteggio con un βcounter. Tutte le prove sono state ripetute tre volte e ogni campione è stato fatto in triplicato 3.6 METODICA UTILIZZATA PER DETERMINARE LA FUNZIONE DEI RECETTORI SULL’ENZIMA ADENILATO CICLASI (3HcAMP) : Il metodo utilizzato per lo svolgimento di questo saggio prevede l’uso di colonne cromatografiche di due tipi secondo il protocollo descritto da AvidorReiss (1995) e Johnson e Salomon (1991). Con questa prova funzionale, si fornisce al sistema cellulare adenina triziata che verrà convertita prima in [ 3H]ATP e quindi in [3H]cAMP attraverso l’enzima adenilato ciclasi. 47 Si valuta esattamente la quantità di cAMP marcato che viene prodotta rispetto al basale (cioè rispetto alle cellule non trattate). Il primo tipo di colonne utillizzato ha come fase stazionaria la resina Dowex 50 (AG 50W – X4 resin, 200400 mesh, hydrogen form BioRad Laboratories) che contiene gruppi solfonici caricati negativamente. Il passaggio attraverso queste colonne garantisce la separazione dei prodotti che vengono dal metabolismo dell’adenina triziata: [3H]ATP, [3H]ADP, il [3H]cAMP. Mentre [3H]ATP e [3H]ADP sono eluiti per volumi di fase mobile minori in quanto hanno rispettivamente tre e due gruppi fosfato carichi negativamente, il [3H]cAMP viene eluito per volumi di fase mobile maggiori. Il secondo tipo di colonne, invece, ha una fase stazionaria di Allumina (Sigma) che non lega il cAMP ma lega gli altri nucleotidi carichi negativamente. Questa caratteristica consente, attraverso opportuni lavaggi con la fase mobile imidazolo 0,1 M, una più accurata separazione del [3H]cAMP dagli altri nucleotidi. Il [3H]cAMP eluito verrà poi raccolto nelle vials dove è stato precedentemente versato il liquido di scintillazione.Ventiquattro ore dopo la transfezione, le cellule sono state tripsinizzate e ridistribuite in piastre da 24 pozzetti contenenti 1 ml di medium di coltura. Dopo 24 h le cellule sono state testate con la prova funzionale dell’adenilato ciclasi. La prova è stata eseguita in triplicato per ogni campione come descritto da AvidorReiss et al. (1995). In breve, dopo aver rimosso il medium, le cellule sono state incubate per 2 h a 37°C con 0.25 ml/pozzetto di medium fresco contenente [3H] adenina (1.25 µL /pozzetto). Successivamente a tale incubazione il medium è stato sostituito con 0.25/pozzetto di DMEM contenente 20 mM HEPES, pH 7.4, due inibitori delle fosfodiesterasi, il 3isobutil1metilxantina (IBMX 0.5 mM) e il RO20 1724 (0.5 mM). L’attività dell’enzima adenilato ciclasi è stata stimolata con forskolina (FS 1 µM) per avere un segnale di base più elevato. L’inibizione dell’attività dell’adenilato ciclasi è stata effettuata tramite stimolazione dei recettori dopaminergici con agonisti quali il quinpirolo e il 7OH DPAT. Dopo 10 min di incubazione a 30°C, il medium è stato rimosso e la reazione è stata terminata aggiungendo acido perclorico al 2.5% contenente cAMP non marcato 0.1 mM e incubando per almeno 30 min a 4°C. L’acido perclorico è stato poi neutralizzato con 100 µl di una soluzione KOH 4.2 M e K2CO3 1.85 M; ogni campione raccolto in apposite eppendorf è stato centrifugato per due min. La quantità di [3H]cAMP formato è stata determinata tramite una procedura di separazione a due passaggi in colonne Dawex e alumina come descritto da AvidorReiss et al. (1995). 48 3.7 STATISTICA: I dati sono stati presentati come media +/ l’errore standard di almeno tre esperimenti. Le curve di saturazione sono state interpolate dall’equazione a= Bmax∗X n K X n (1) per derivare il coefficiente di Hill (n) e dall’equazione a= Bmax∗X K d X (2) per ottenere la costante di dissociazione Kd e la capacità massima di binding Bmax. I valori di a e di X nelle due equazioni precedenti rappresentano rispettivamente la quantità di [3H]nemonapride legato specificatamente al tessuto e la concentrazione dello stesso non legato. Il coefficiente K nell’equazione (1) rappresenta una costante generica che per n = 1 coincide con la Kd. I dati degli esperimenti di inibizione sono stati interpolati usando l’equazione a=100− 100∗X n K X n (3) per calcolare il coefficiente di Hill (n), e l’equazione a=100− 100∗X IC 50 X (4) 49 per calcolare il valore di IC50. La IC50 rappresenta la concentrazione dell’inibitore che sposta il 50% del radioattivo legato specificatamente ai recettori. I valori di IC50 sono stati trasformati in Ki con la formula di Cheng e Prusoff (1973) che è la seguente: K i= IC 50 1F / K d (5) dove F rappresenta la concentrazione della [3H]nemonapride usata nell’esperimento d’inibizione. Quando il coefficiente di Hill era diverso da 1, come nel caso di tutti gli agonisti, il valore di IC50 è stato trasformato in IC50 corretto (IC50 corr) grazie alla formula di Cheng e Prusoff. Il programma usato per calcolare tutte le costanti è stato ORIGIN con un computer IBMcompatibile. 50 4. RISULTATI 4.1 AFFINITA' DEI VARI LIGANDI PER I RECETTORI DOPAMINERGICI D3 E D2 . In studi di spiazzamento effettuati con [3H]Nemonapride, aripiprazolo, S33592, bifeprunox, Ndesmetilclozapina e preclamolo hanno dimostrato di possedere una affinità significativa sia per il D2 che per il D3, sebbene con marcata differenza nella loro potenza (Tabella 1). S33592, NDMC e preclamolo hanno una affinità lievemente maggiore per il D2 rispetto al D3 mentre l’aripiprazolo ha una preferenza 10 volte maggiore per il D2. Si osserva una situazione contraria per il bifeprunox che si lega preferenzialmente, anche se in modo modesto, al D3. E' stato quindi osservato che Aripiprazolo e Bifepronux sono i ligandi più potenti rispettivamente per il recettore D2 e D3, mentre il Preclamolo è il ligando più debole per entrambi i recettori. Aripiprazole Bifeprunox NDMC S33592 Preclamol D2 (Ki, nM) D2trunk/D3tail (Ki, nM) D3trunk/D2tail (Ki, nM) D3 (Ki, nM) 0.74 ± 0.007 2.26 ± 0.13 73.4 ± 12.1 28.6 ± 2.84 273 ± 22.4 14.3 ± 1.79 1.25 ± 0.12 88.6 ± 15.3 227 ± 15.8 1,532 ± 209 1.38 ± 0.13 0.67 ± 0.06 36.6 ± 6.29 10.6 ± 1.12 133 ± 14.9 7.61 ± 1.27 0.73 ± 0.29 167 ± 15.9 77.4 ± 6.8 702 ± 81.4 Tabella 1: Profili di Binding degli antipsicotici sui recettori D2 e D3 wildtype e sui recettori chimerici tronchi D2trunkD3tail e D3trunkD2tail. 4.2 INFLUENZA DEGLI ANTIPSICOTICI SULL’ATTIVITA' DELL’ADENILATO CICLASI V/VI STIMOLATA CON FORSKOLIN IN CELLULE COS7 TRANSFETTATE CON I RECETTORI D2 O D3. L’incremento di cAMP sopra i livelli basali dopo la stimolazione con forskolin della adenilato ciclasi endogena contenuta nelle cellule COS7 è in media di solo 2 volte. Per poter amplificare la sensibilità del sistema, in tutti gli esperimenti funzionali eseguiti in cellule COS7 abbiamo cotransfettato una adenilato ciclasi chimerica ACV/VI. Con questa adenilato ciclasi, l’incremento medio di cAMP sopra i livelli basali è stato di circa 7 volte (Scarselli et al., 2001; Robinson and Caron, 1997). Tutti gli agonisti testati hanno mostrato verso il recettore D3 un’attività nulla (Tabella 2). Al contrario tutti hanno mostrato di inibire l’adenilato ciclasi in presenza del recettore D2. Il Quinpirolo si comporta come un agonista pieno inibendo fortemente l’attività dell’Adenilato ciclasi V/VI stimolato con forskolin (Tabella 2 e F1g. 1). Tutti gli altri composti, preclamolo, bifeprunox, NMDC, aripirpazolo e S33592 si sono comportati invece come agonisti parziali nel sopprimere 51 l’attività dell’enzima indotto dalla forskolina, con un range di inibizione che andava dal 16,3 % per il preclamolo fino al 23 % per il Bifeprunox (Tabella 2 e Fig. 2, 3, 4, 5 e 6). L’Aripiprazolo ed il Preclamolo sono risultati essere i ligandi rispettivamente più potente e meno potente nello stimolare il recettore D2. D2L + ACV/VI Quinpirole Aripiprazole D2L + D3 + ACV/VI D3 + ACV/VI IC50 (nM) IC50 (nM) IC50 (nM) (Emax, %) 2.1 ± 0.5 ( Emax, %) 1.47 ± 0.71 ( Emax, %) (48.4 ± 1.89) 4.46 ± 0.73 (43.2 ± 3.33) No inhibition No inhibition No inhibition No inhibition No inhibition No inhibition No inhibition No inhibition No inhibition No inhibition No inhibition (18.9 ± 0.53) 21.5 ± 4.3 Bifeprunox NDMC (23.3 ± 0.94) 173 ± 35.3 (22.8 ± 0.92) 16.7 ± 8.8 S 33592 Preclamol (18.1 ± 1.89) 97.4 ± 40.6 (16.3 ± 1.29) Tabella 2: Influenza degli antipsicotici sull’attività dell’adenilato ciclasi V/VI indotta con Forskolina. 52 cAMP accumulation (% of FKstimulated) 110 100 90 80 70 D2L + D3 D2L 60 50 11 0 ,0 1 0 ,1 10 91 8 10 7 100 6 1000 Quinpirole log[M] Figura 1: Effetto inibitorio del quinpirolo sull’attività dell’AC V/VI stimolata con forscolina in cellule COS 7 transfettate con il solo recettore D2L o cotransfettate con entrambi i recettori D2L e D3. Tutti i farmaci sono stati poi testati su cellule cotransfettate con i recettori D2 e D3. Il Quinpirolo ha avuto un potente effetto inibitorio sull’AC V/VI con una potenza paragonabile a quella osservata nelle cellule transfettate con il solo recettore D2 (Tabella 2 e Fig. 1). Questo effetto sui recettori D2 e D3 cotransfettati è stato osservato anche con altri agoisti pieni quali il Pergolide, il pramipexolo, il Ropinirolo e lo S32504 ( Maggio et al, 2003 ) (Table 3). 53 D2L + ACV/VI D2L + D3 + ACV/VI IC50 (nM) [Emax, %] IC50 (nM) [Emax, %] S 32504 49.5 [38.2] 1.50 [40.9] Pramipexolo 21.5 [43.3] 1.98 [52] Ropinirole 75.5 [33] 3.89 [49.4] Pergolide 7.86 [30.2] 4.31 [40.7] Tabella 3 : Influenza di alcuni agonisti dopaminergici sull’attività dell’AC V/VI In cellule COS 7 cotransfettate con i recettori D2 e D3 e su cellule COS 7 transfettate e con il solo D2 Aripiprazolo, S33592, Bifepronux, NMDC e Preclamolo invece non hanno inibito l’attività dell’AC V/VI in cellule transfettate con D2 e D3 questo in contrasto con le loro proprietà di agonisti parziali in cellule transfettate con il solo recettore D2 (Tabella 2 e Fig. 2, 3, 4, 5 e 6). cAMP accumulation (% of FKstimulated) 110 100 90 D2L + D3 D2L 80 70 110 ,0 1 100 ,1 9 1 8 10 71 0 0 61 0 0 0 Aripiprazole log[M] 54 Figura 2 : effetto dell’aripiprazolo rispettivamente su cellule COS 7 transfettate con il recettore D2 e su celluleCOS 7 cotransfettate con entrambi i recettori D2 e D3. cAMP accumulation (% of FKstimulated) 110 100 90 D2L + D3 D2L 80 70 11 0 ,0 1 0 ,1 10 91 8 10 7 100 6 1000 S33592 log[M] Figura 3 : effetto dell’S33592 rispettivamente su cellule COS 7 transfettate con il recettore D2 e su celluleCOS 7 cotransfettate con entrambi i recettori D2 e D3. 55 cAMP accumulation (% of FKstimulated) 110 100 90 D2L + D3 D2L 80 70 11 0 ,1 1 10 9 10 8 100 7 1000 6 10000 Preclamol log[M] Figura 4 : effetto del Preclamolo rispettivamente su cellule COS 7 transfettate con il recettore D2 e su celluleCOS 7 cotransfettate con entrambi i recettor D2 e D3. 56 cAMP accumulation (% of FKstimulated) 110 100 90 D2L + D3 D2L 80 70 11 0 ,1 1 10 9 10 8 100 7 1000 6 10000 NDesmethyl Clozapine log[M] Figura 5 : effetto dell’NDesmetil Clozapina rispettivamente su cellule COS 7 transfettate con il recettore D2 e su celluleCOS 7 cotransfettate con entrambi i recettori D2 e D3. 57 4.3 INFLUENZA DEGLI ANTIPSICOTICI SULL’ATTIVITA' DELL’AC V/VI STIMOLATA CON FOESKOLINA IN CELLULE COS7 COTRANSFETTATE CON I RECETTORI D2 E D3 E TRATTATE CON QUINPIROLO. Come descritto sopra, il quinpirolo inibisce l’attività dell’AC V/VI stimolata con Forskolina sia in cellule transfettate solamente con D2 che in cellule cotransfettate con D2 e D3. Alla concentrazione di 10 nM l’inibizione che si ottiene è di circa 40 %. Coincubando l’Aripiprazolo con il quinpirolo 10 nM in cellule transfettate con il solo recettore D2 si è avuta una parziale riduzione dell’effetto inibitorio del quinpirolo senza però avere una completa abolizione dell’inibizione. In pratica l’aripiprazolo comportandosi da agonista parziale ha prima ridotto l’effetto del quinpirolo e a saturazione ha stimolato parzialmente il recettore D2 (Fig. 6). 110 D2L D2L + D3 90 80 Quinpirole 10 nM cAMP accumulation [% FKstimulated] 100 70 60 50 0 ,0 1 010 ,1 19 18 0 100 7 Aripiprazole log[M] 1 06 00 58 Figura 6 : Riduzione dell’effetto inibitorio del quinpirolo in cellule transfettate con il solo recettore D2 e in cellule cotransfettate con i recettori D2 e D3 da parte dell’aripiprazolo. In cellule cotransfettate con entrambi i recettori D2 e D3, gli effetti inibitori del quinpirolo sono stati aboliti dall’aripiprazolo. Questo è dovuto al fatto che come osservato nel paragrafo precedente, l’aripiprazolo sui recettori D2 e D3 cotransfettati si comporta come un antagonista invece che come un agonista parziale. Effetti simili si sono osservati con il composto S33592. Quindi anche con questo composto, in cellule transfettate con il solo D2 si è avuta una inibizione parziale dell’effetto inibitorio del quinpirolo, mentre in cellule cotransfettate con D2 e D3 il S33592 ha completamente inibito l’effetto del quinpirolo (Fig. 7). 110 D2L D2L + D3 90 80 Quinpirole 10 nM cAMP accumulation [% FKstimulated] 100 70 60 50 0 ,0 1 10 0 ,1 9 8 S 33592 log[M] 1 10 100 7 6 1000 59 Figura 7 : Inibizione da parte dell’S33592 dell’effetto inibitorio del Quinpirolo su cellule transfettate con il solo recettore D2 e su cellule cotransfettate con i recettori D2 e D3. 60 4.4 INFLUENZA DEGLI ANTIPSICOTICI SULL’ATTIVITA' DELL’ADENILATO CICLASI V/VI STIMOLATA CON FORSKOLINA IN CELLULE COS7 TRANSFETTATE CON I RECETTORI CHIMERICI FRAMMENTATI D2TRUNK/D3TAIL E D3TRUNK/D2TAIL In cellule cotransfettate con i frammenti trunk e tail dei recettori dopaminergici D2 e D3: D2TRUNK e D3TAIL O D3TRUNK eD2TAIL, il quinpirolo ha inibito marcatamente l’attività dell’ AC V/VI stimolata con Forskolina. Al contrario del quinpirolo, aripiprazolo, S33592, bifeprunox e NMDC non hanno inibito l’attività dell’ AC V/VI stimolata con Forskolina in cellule cotransfettate con i frammenti recettoriali D2TRUNK/D3TAIL O D3TRUNK/D2TAIL (Fig.8). 61 Inoltre tutti questi composti alla concentrazione di 100 nM hanno inibito l’effetto agonista del quinpirolo 10 nM comportandosi da antagonisti (fig.9). 120 D2trunk/D3tail D3trunk/D2tail 100 90 cAMP accumulation (% of FSstimulated) 110 80 70 60 FK Quinp. Aripip. S33592 Precla. Bifep. NDMC 62 figura 8: Attività del quinpirolo e degli antipsicotici su cellule COS 7 cotransfettate con i recettori D2TRUNK e D3TAIL o D3TRUNK eD2TAIL. 120 D2trunk/D3tail D3trunk/D2tail 100 90 cAMP accumulation (% of FSstimulated) 110 80 70 60 Cont. Quin. Quin. + Quin. + Quin. + Quin. + Quin. + Aripip. S33592 Precla. Bifep. NDMC figura 9: Attività degli antipsicotici alla concentrazione di 100 nM coincubati con quinpirolo 10 nM su cellule COS 7 cotransfettate con i recettori D2TRUNK e D3TAIL o D3TRUNK eD2TAIL. 4.5 INFLUENZA DEGLI ANTIPSICOTICI SULL’ATTIVITA' DELL’ADENILATO CICLASI V/VI STIMOLATA CON FORSKOLINA IN CELLULE COS7 TRANSFETTATE CON IL RECETTORE D2L ED IL RECETTORE CHIMERICO D3i3(D2) In questa serie di studi abbiamo impiegato un recettore D3 modificato , il D3i3(D2) in cui il segmento carbossiterminale del terzo loop citoplasmatico è stato scambiato con la sezione equivalente del recettore D2l. 63 Questa alterazione incrementa marcatamente l’efficacia di accoppiamento del recettore D3 alla proteina G, senza però alterare il profilo di binding del recettore wild type (Filteau et al,1999 ). Nelle cellule transfettate col recettore D3i3(D2), in contrasto al recettore D3 wild type, il quinpirolo sopprime l’attività dell’ AC V/VI stimolata con Forskolina con un’efficacia del 35 % e questo effetto è simile a quello ottenuto sul recettore D2l (Tabella 4 e Fig. 10). D2L + ACV/VI D2L + D3/D2i3 + ACV/VI D3/D2i3 + ACV/VI IC50 (nM) IC50 (nM) IC50 (nM) (Emax, %) 1.4 ± 0.25 ( Emax, %) 1.5 ± 0.21 ( Emax, %) 1.6 ± 0.45 (41.4 ± 1.0) 3.6 ± 1.9 (45.3 ± 1.3) 4.6 ± 1.4 (34.6 ± 0.98) 2.2 ± 0.78 (19.3 ± 1.4) 12.1 ± 4.05 (24.8 ± 1.1) 14.1 ± 1.11 (18.7 ± 0.9) 21.7 ± 4.45 (18.8 ± 1.22) (21.4 ± 0.33) (19.6 ± 0.8) Quinpirole Aripiprazole S33592 Tabella 4: Influenza degli antipsicotici sull’attivita' dell’adenilato ciclasi V/VI stimolata con forskolina in cellule transfettate con il recettore D2l ed il recettore chimerico D3i3(D2) 64 110 cAMP accumulation (% of FKstimulation) 100 90 80 70 D2L D2L + D3i3D2 D3i3D2 60 50 11 0 ,0 1 0 ,1 10 91 8 10 7 100 6 1000 Quinpirole log[M] Figura 10: Attività del Quinpirolo sull’attivita' dell’adenilato ciclasi V/VI stimolata con forskolina in cellule COS 7 cotransfettate con il recettore D2l ed il recettore chimerico D3i3(D2), in cellule transfettate con il D3i3(D2) ed in cellule transfettate con il D2. Anche l’Aripiprazolo e l’S33592 su questo recettore mutato inducono una marcata riduzione dell’incremento dei livelli di cAMP (Tabella 4 e Fig. 12 e 13). 65 cAMP accumulation (% of FKstimulation) 110 100 90 D2L D2L + D3i3D2 D3i3D2 80 70 11 0 ,0 1 91 0 ,1 10 8 10 6 7 100 1000 Aripiprazole log[M] Figura 11: Attività dell’ Aripiprazolo sull’attivita' dell’adenilato ciclasi V/VI stimolata con forskolina in cellule COS 7 cotransfettate con il recettore D2l ed il recettore chimerico D3i3(D2), in cellule transfettate con il D3i3(D2) ed in cellule transfettate con il D2. cAMP accumulation (% of FKstimulation) 110 100 90 D2L D2L + D3i3D2 D3i3D2 80 70 110 ,0 1 100 ,1 9 1 8 10 71 0 0 61 0 0 0 S 33592 log[M] 66 Figura 12: Attività dell’S33592 sull’attivita' dell’adenilato ciclasi V/VI stimolata con forskolina in cellule COS 7 cotransfettate con il recettore D2l ed il recettore chimerico D3i3(D2), in cellule transfettate con il D3i3(D2) ed in cellule transfettate con il D2. Lo stesso effetto dei tre agonisti si è osservato in cellule cotransfettate con i recettori D2l e D3i3(D2) (Tabella 4 e Fig. 11, 12 e 13). Quindi la potenza e l’efficacia dei tre ligandi sono comparabili sia in cellule transfettate con i soli D2l o D3i3D2 e sia in cellule cotransfettate con entrambi i recettori. Anche Bifeprunox, NDMC e Preclamolo hanno mostrato la stessa attività inibitoria sull’AC V/VI in cellule trasfettate o co transfettate con i recettori D2l e D3i3(D2) (Tabella 4 e Fig. 13). Control Quinpirole Aripip. S33592 Preclamol Bifeprunox NDMC 100 cAMP accumulation (% FKstimulated) 120 80 60 D2L D2L + D3i3D2 D3i3D2 67 Figura 13: influenza degli antipsicotici sull’attivita' dell’adenilato ciclasi v/vi stimolata con forskolina in cellule COS 7 cotransfettate con il recettore D2l ed il recettore chimerico D3i3(D2) e in cellule transfettate con il solo D2l o con il solo D3i3(D2). 68 4.6 ESPERIMENTI SU CELLULE CHO STABILMENTE TRANSFETTATE CON I RECETTORI D2 E D3 Alcuni esperimenti sono stati ripetuti su cellue stabilmente transfettate con i soli recettori D2 e D3 e cotransfettate con ambedue i recettori. Il motivo per il quale si sono ripetuti alcuni esperimenti su queste cellule è dovuto al fatto che l’espressione recettoriale in queste cellule è stabile e non varia da trasfezione a trasfezione. In questo sistema cellulare abbiamo provato l’attività agonista parziale dell’aripiprazolo e del S33592. Le Fig. 15 e 16 dimostrano chiaramente che anche in queste cellule l’effetto agonista parziale è presente solo nelle cellule CHOD2L mentre non è presente nelle cellule CHOD3 e CHOD2L/D3. cAMP accumulation (% of FKstimulation) 110 100 90 D2L D2L + D3 D3 80 70 110 ,0 1 100 ,1 9 1 8 10 71 0 0 61 0 0 0 Aripiprazole log[M] Figura14: Effetto dell’Aripiprazolo su cellule CHO transfettate stabilmente con i recettori D2l o D3 o cotransfettate stabilmente con entrambi i recettori. 69 cAMP accumulation (% of FKstimulation) 110 100 90 D2L D2L + D3 D3 80 70 110 ,0 1 100 ,1 9 1 8 10 71 0 0 61 0 0 0 S 33592 log[M] Figura 15:Effetto dell’S33592 su cellule CHO transfettate stabilmente con i recettori D2l o D3 o cotransfettate stabilmente con entrambi i recettori. 70 5. DISCUSSIONE In lavori precedenti (Scarselli et al,2001: Maggio et al,2003) è stato osservato come S32504, Pramipexolo, Ropinirolo e 7 OHDPAT, agonisti per i recettori D2, inibiscano l’attività dell’ AC V/VI stimolata con Forskolina con maggiore potenza in cellule cotrasfettate con i recettori D2l e D3. Essendo questa isoforma di AC non stimolabile dal recettore D3, questo incremento della potenza degli agonisti è stato interpretato con la formazione di eterodimeri D2l/D3. In questo lavoro, al contrario, alcuni agonisti parziali per il recettore D2 come Aripiprazolo, S33592, Bifeprunox, N Desmetilclozapina e Preclamolo hanno dato risultati differenti, si è osservato infatti un mancato effetto di questi composti sulle cellule cotransfettate con i recettori D2 e D3. 5.1 PROFILI DI BINDING DEGLI ANTIPSICOTICI SUI RECETTORI D2 E D3 : confronto tra le affinità degli antipsicotici per i siti recettoriali D2l e D3 L’aripiprazolo ha mostrato di possedere una forte affinità per il recettore D2l , mentre la sua affinità per i siti D3 è circa dieci volte più bassa, un valore che coincide con l’affinità valutata precedentemente (9.0 e 9.1 nM). L’aripiprazolo mostra quindi di avere preferenza per il sito D2l rispetto al D3. Il Bifeprunox ha mostrato una maggiore affinità per il D3 rispetto al D2L. Per quanto riguarda l’NMDC, sono state osservate in precedenti studi (Burnstein et al,2005) affinità di 89 e 153 nM rispettivamente per i siti hD2s e D3 espressi in cellule NIH737 . Nel presente studio sono stati osservasti dati in linea con quelli ottenuti da Burnstein, infatti, le affinità osservate sono di 73 e 153 nM rispettivamente per i siti hD2l e D3. Per il Preclamolo l’affinità per il recettore hD2l (in cellule CHO) è di norma compresa tra 300 e 1000 nM e il valore che abbiamo ottenuto è di 273 nM , in accordo con questo range. Inoltre l’affinità del preclamolo per il sito D3 è comparabile con quella ottenuta da Chio et al (1993) impiegando [3H]Spiroperidolo (330nM). Infine l’affinità dell’S33592 per il sito D2l (29 nM), è simile a quella osservata per tale sito espresso da cellule CHO (39nM) (Gobert et al,2000). Utilizzando [3H]Spiperone, S33592 è più potente sul sito hD3 (11nM) (Gobert et al,2000) di quanto osservato in questo lavoro e la ragione non è ancora chiara. 71 5.2 AZIONE DEGLI ANTIPSICOTICI SUL RECETTORE D2L L’azione dell’aripiprazolo sui recettori D2L e D2s dipende da diversi fattori quali il segnale intracellulare, il sistema di espressione, la densità dei recettori e altri fattori (Lawler et al,1999;Burris et al,2002;Shapiro et al,2003;Aihara et al,2004;Burstein et al,2005;Tadori et al,2005). I nostri dati estendono queste osservazioni mostrando come l’aripiprazolo si comporti come agonista parziale relativamente al quinpirolo in cellule COS 7 che esprimono il recettore D2. Per il Bifeprunox non sono disponibili molte informazioni del suo profilo farmacologico, comunque è stata documentata un azione di agonista parziale sulla ciclasi, in cellule CHO transfettate con il D2, e un’azione di agonista parziale sempre sul recettore D2 per la stimolazione delle MAP Kinasi (Van Vliet et al,2000; Bruins Slot et al, 2006). Il Bifeprunox sembra essere molto più efficace dell’aripiprazolo come agonista parziale, questa osservazione è comparabile con i nostri risultati dove il Bifeprunox sembra essere molto più efficiente dell’aripiprazolo nel l’inibire l’AC V/VI cotransfettata con il recettore D2L. Un recente studio ha dimostrato le proprietà di agonista parziale dell’NMDC sui recettori hD2s costitutivamente attivati in cellule NIH3T3 in cui la proteina Gαo era stata sovraespressa (Burstein et al, 2005). Inpiegando un protocollo diverso, nel presente studio abbiamo approfondito l’azione di agonista parziale dell’NMDC sul recettore D2. L’NMDC nel nostro studi è risultato essere un agonista parziale ma in accorso allo studio precedente la sua potenza è risultata minore di quella dell’aripiprazolo. Sempre in analogia con studi precedenti il nostro studio ha mostrato che l’agonista parziale Preclamolo era meno potente dell’agonista parziale aripiprazolo. Il presente studio tuttavia è il solo, a differente di quelli fatti in precedenza, che paragona direttamente aripiprazolo, bifeprunox, NMDC e preclamolo, e dimostra la loro azione di agonisti parziali per i recettori D2. Il nostro studio ha inoltre compreso il composto S33592. Questo ligando è stato precedentemente studiato per il suo effetto di agonista parziale per il recettore D2 accoppiati a Gi/o e per la MAPKinasi. Inoltre è stato studiato il suo meccanismo di agonista parziale sui recettori D2 cerebrali che controllano il rilascio di prolattina (Gobert et al,2000). 72 5.3 PROFILI DI BINDING DEGLI ANTIPSICOTICI TESTATI SUI RECETTORI CHIMERICI D2TRUNK/D3TAIL E D3TRUNK/D2TAIL I profili farmacologici degli antipsicotici sui recettori frammentati chimerici D2trunk/D3tail e D3trunk/D2tail sono stati considerati in parallelo con i profili farmacologici dei recettori D2L e D3. Le affinità di Aripiprazolo e Bifeprunox sono risultate maggiori rispetto a S33592 e NMDC e acnor di più rispetto al Preclamolo che è stato il farmaco che ha mostrato una minore potenza. Le attività di questi farmaci sui recettori chimerici frammentati suggeriscono che essi potrebbero svolgere una funzione su eterodimeri D2/D3 formati in seguito al meccanismo di domain swapping tra i recettori D2 e D3. 5.4 AZIONI FUNZIONALI DEGLI ANTIPSICOTICI SUI RCETTORI D2L E D3 COTRANSFETTATI E SUI RECETTORI CHIMERICI FRAMMENTATI D2TRUNK/D3TAIL E D3TRUNK/D2TAIL S33592, aripiprazolo, bifeprunox, NMDC e preclamolo sono risultati tutti inattivi in cellule cotransfettate con i recettori D2L e D3. Questa mancata funzione è stata osservata sia in cellule COS 7 trasfettate transientemente che in cellule CHO transfettate stabilmente. Inoltre gli stessi composti hanno mostrato una mancata attività a livello dei recettori chimerici frammentati D2trunk/D3tail e D3trunk/D2tail. Siccome sia l’aripiprazolo che gli altri agonisti perdono efficacia funzionale in cellule cotransfettate con i recettori D2 e D3, sembrerebbe che la maggior parte dei recettori D2, venga reclutata dai recettori D3 nella formazione di eterodimeri. 5.5 AZIONE DEGLI ANTIPSICOTICI SU CELLULE TRANSFETTATE CON IL RECETTORE CHIMERICO D3i3(D2) E SU CELLULE COTRANSFETTATE CON IL RECETTORE D2L E CON IL D3i3(D2) Nel recettore chimerico D3i3(D2), 12 aminoacidi all’estremità carbossi terminale del recettore D3 sono sostituiti con la corrispondente sequenza aminoacidica del recettore D2. Questo mutante così costruito mantiene il profilo farmacologico del recettore D3 wildtype, ma contrariamente a quest’ultimo è funzionalmente attivo. In accordo con questa sua capacità funzionale, Aripiprazolo, S33592, Bifepronux, NMDC, e Preclamolo, si comportano su questo recettore come agonisti parziali. 73 Chiaramente questi ligandi possiedono una significativa, sebbene submassimale, attività intrinseca anche sui recettori D3 anche se questa non è statamessa in evidenza nel nostro sistema sperimentale in quanto la capacità di accoppiamento alle proteine G del D3 è nel nostro caso inesistete. Burstein et al(2005) hanno riportato le proprietà di agonista parziale dell’aripiprazolo per il D3 in cellule che sovraesprimono la isoforma della proteina G, Gα/o. Le proprietà di agonista parziale dell’aripiprazolo sul D3 sono state osservate anche in uno studio di Shapiro et al, (2003) studiando l’aperura dei canali al potassio. Inoltre, l’aripiprazolo ha mostrato attività di agonista parziale sui recettori D3 misurando la fosforilazione di MAPKinasi (Gobert et al,2000). Non sono disponibili in letteratura dati che riguardino il bifeprunox, esso comunque attiva in modo submassimale il recettore hD3 accoppiato alle MAPKinasi ( Gobert et al,2000; Mannouri la Cour C and Millan M.J., unpub.obs.) . Anche NMDC si comporta come agonista parziale sul recettore D3 accoppiato all’ AcII ( Shapiro et al,2003), mentre sono documentate le azioni di agonista parziale del preclamolo sul recettore hD3 accoppiato all’attivazione dell’adenilato ciclasi e alla mitogenesi. Sulle cellule cotransfettate con i recettori D3i3(D2) e D2L, Aripiprazolo, S33592, bifeprunox, NMDC e preclamolo si comportano tutti come agonisti parziali e sopprimono l’attività dell’ AC V/VI stimolata con Forskolina. Inoltre, analogamente al quinpirolo, sia l’aripiprazolo che l’S33592 hanno mostrato un effetto inibitorio sulle cellule cotransfettate con D3i3(D2) e D2L. Questi dati ottenuti utilizzando il recettore D3i3D2 suggeriscono che il mancato effetto di questi agonisti parziali che abbiamo osservato sul recettore D3 dipende dal fatto che questo recettore è debolmente accoppiato alle proteine G e quindi nel nostro sistema non era possibile rilevarne la sua funzione. 74 5.6 POTENZIALE AZIONE DEGLI ANTIPSICOTICI SUGLI ETERODIMERI D3/D2 NELL’ENCEFALO Gli autorecettori D3 e D2s colocalizzati nei neuroni dopaminergici controllano la sintesi ed il rilascio fasico e tonico della dopamina , interagendo con la tiroxina idrossilasi , con il meccanismo di liberazione della dopamina e con i trasportatori della dopamina, anche se il loro rispettivo ruolo rimane indefinito. Siccome i recettori presinaptici D2 e D3 sono molto sensibili, l’effetto di ogni agonista parziale sugli eterodimeri D3/D2 è difficilmente definibile. Infatti Aripiprazolo, preclamolo, S33592, bifeprunox e NMDC generalmente attenuano o non modificano l’attività dei neuroni dopaminergici nel ratto. Sebbene sia stato riscontrato che l’aripiprazolo incrementa il rilascio di dopamina nella corteccia prefrontale e nell’ippocampo ( Li et al,2004), questa azione riflette principalmente il reclutamento di recettori 5Ht1A (Shapiro et al,2003; Newman Tancredi et al, 2005; Bruins Slot et al,2006). Analogamente l’elevato rilascio di dopamina causato da NMDC riflette largamente la stimolazione dei recettori muscarinici M1 ( Li et al,2005) . Il blocco degli eterodimeri degli autorecettori D2/D3, può essere interessante in certe condizioni. I recettori D3 postsinaptici dei gangli basali sono espressi ad alte concentrazioni nei neuroni positivi per la sostanza P che esprimono anche recettori D1 piuttosto che nei neuroni positivi per l’enkefalina che esprimono i recettori D2. La somministrazione di LDopa cronicamente induce l’espressione dei recettori striatali D3 ( Bordet et al,1997 and 2000; Morisette et al,1998; Quik et al,2000). Inoltre, insieme ai recettori D2, i recettori D3 sono molto rappresentati nel Nucleo Accumbens e in altre strutture del sistema libico, e la loro densità è maggiore nei primati rispetto ai roditori (MeadorWoodruff et al, 1996; Morisette et al, 1998; Diaz et al, 2000; Quik et al, 2000; Stanwood et al, 2000; Joyce, 2001). Tutte queste considerazioni indicano che la probabilità di formazione degli eterodimeri D2 e D3 è maggiore nei primati rispetto ai roditori. Nell’uomo un aumento nella densità dei recettori D3 mesolimbici è associato alla schizofrenia e all’abuso di cocaina (Staley and Mash, 1996; Gurevich and Joyce, 1997; Segal et al, 1997; Joyce, 2001). I modelli sperimentali nei roditori, e nei primati, sono i mezzi migliori per dimostrare l’importanza della formazione di eterodimeri D2 e D3 nell’encefalo e il loro ruolo funzionale. Aripiprazolo, bifeprunox, ed NMDC esercitano la loro azione anche sui recettori 5HT1A e su altre classi di recettori in 75 vivo, complicando così l’interpretazione della loro azione (Shapiro et al, 2003). Di conseguenza S33592, che mostra grande selettività per i recettori D2 e D3 (Gobert et al, 2000; Rivet et al, 2000) può essere lo strumento sperimentale più appropriato per indagare sul significato degli eterodimeri D2/D3 nel Sistema Nervoso Centrale. 76 6. CONCLUSIONI Nell’insieme questi dati dimostrano che aripiprazolo, bifeprunox, NMDC, preclamolo e S33592 si comportano come agonisti parziali nei confronti dei recettori dopaminergici D2. Al contrario questi antipsicotici si comportano come antagonisti quando il recettore D2 viene espresso insieme al recettore D3, e questo è dovuto probabilmente alla loro formazione di eterodimeri. Sebbene non sia possibile dedurre la precisa azione dei farmaci su popolazioni discrete di recettori D2/D3 nell’encefalo, queste scoperte hanno importanti implicazioni. Quindi l’azione degli antipsicotici come agonisti parziali potrebbe non riflettere una bassa efficacia nella stimolazione dei recettori D2 e/o D3 ma, piuttosto, in analogia con altri antipsicotici, potrebbe riflettere il blocco degli eterodimeri D2/D3 (e/o dei recettori D3) postsinaptici. Questa intrigante ipotesi giustifica ulteriori indagini. 77 7. BIBLIOGRAFIA AbdAlla S., Lother H., Massiery A. and Quitterer U. (2001) Increased AT1 receptor heterodimers in preeclampsia mediate enhanced angiotensin II responsiveness. Nat. Med. 7 (9), 10031009. Acquas E., Tanda G. and Di Chiara G. (2002) Differential effects of caffeine on dopamine and acetylcholine transmission in brain areas of drugnaive and caffeinepretreated rats. Neuropsychopharmacology 27, 182193. Agnati LF, Ferre S, Lluis C, Franco R, Fuxe K (2003). Molecular mechanisms and therapeutical implications of intramembrane receptor . receptor interaction among heptahelical receptors with examples from striatopallidal GABA neurons. Pharmacol Rev 55: 509–550. Aihara K, Shimada J, Miwa T, Tottori K, Burris KD, Yocca FD et al (2004). The novel antipsychotic aripiprazole is a partial agonist at short and long isoforms of D2 receptors linked to the regulation of adenylyl cyclase activity and prolactin release. Brain Res 1003: 917. Asghari V., Schoots O., Van Kats S., Ohara K., Jovanovic V., Guan H.C., Bunzow J.R., Petronis A., and Van Tol H.H.M. (1994) Dopamine D4 receptor repeat: analysis of different native and mutants forms of the human and rat genes. Mol. Pharmacol. 46, 364373. AvidorReiss T., Bayewitch M., Levy R., MatusLeibovitch N., Nevo I. and Vogel Z. (1995) Adenylyl cyclase supersensitization in opioid receptortransfected Chinese hamster ovary cells following chronic opioid treatment. J. Biol. Chem. 270, 2973229738. Bai M., Trivedi S. and Brown E.M., (1998) Dimerization of the extracellularcalciumsensing receptor (Ca++R) on the cell surface of Ca++Rtransfected HEK 293 cells. J. Biol. Chem. 273, 2360523610. Bassareo V. and Di Chiara G. (1999) Modulation of feelinginduced activation of mesolimbic dopamina transmission by appetitive stimuli and its relation to motivational state. Eur. J. Neuro. 11, 43894397. 78 Bordet R, Ridray S, Carboni S, Diaz J, Sokoloff R, Schwartz JC (1997). Induction of dopamine D3 receptor expression as a mechanism of behavioral sensitization to Levodopa. Proc Natl Acad Sci USA 9: 33633367. Bordet R, Ridray S, Schwartz JC, Sokoloff P (2000). Involvement of the direct striatonigral pathway in levodopainduced sensitization in 6hydroxydopaminelesioned rats. Eur J Neurosci 12: 21172123. Bertorello A.M., Hopfield J.F., Aperia A. and Greengard P. (1990) Inhibition by dopamine of (Na +/K+) ATPasi activity in neostriatal neurons through D1 and D2 receptor synergism. Nature 347, 386388. Bockaert J. and Pin J. (1999) Molecular tinkering of G proteincoupled receptors: An evolutionary success. EMBO J. 18 (7), 17231729. Bouvier M. (2001) Oligomerization of Gproteincoupled transmitter receptors. Nat. Rev. Neurosci. 2 (4), 274286. Bruins Slot LA, De Vries L, NewmanTancredi A, Cussac D (2006). Differential profile of antipsychotics at serotonin 5HT1A and dopamine D2S receptors coupled to extracellular signalregulated kinase. Eur J Pharmacol, in press. Burris KD, Molski T, Xu C, Ryan E, Tottori K, Kikuchi T et al (2002). Aripiprazole, a novel antipsychotic, is a high affinity partial agonist at human dopamine D2 receptors. J Pharmacol Exp Ther 302: 381389. Burstein E.S., Spalding T.A., and Brann M.R. (1998) The second intracellular loop of the M5 muscarinic receptor is the switch which enable a Gprotein coupling. J. Biol. Chem. 273, 2432224327. Burnstein ES, Ma J, Wong S, Gao Y, Pham E, Knapp AE et al (2005). Intrinsic efficacy of antipsychotics at human D2, D3, and D4 dopamine receptors: identification of the clozapine metabolite Ndesmethylclozapine as a D2/D3 partial agonist. J Pharmacol Exp Ther 315: 12781287. Butkerait P., Zheng Y. and Hallak H. (1995) Expression of the human 5hydroxytriptamine A 1 receptor in Sf9 cells: reconstitution of a coupled phenotype by coexpression of a mammalian G proteins subunits. J. Biol. Chem. 270, 18691 79 18699. Cai G., Zhen X., Uryu K and Friedman E. (2000) Activation of Extracellular SignalRegulated Protein Kinases Is Associated with a Sensitized Locomotor Response to D2 Dopamine Receptor Stimulation in Unilateral 6 HydroxydopamineLesioned Rats. J. Neuro. 20 (5), 18491857. Calabresi P., Maj R., Mercuri N.B. and Bernardi G. (1992) Coactivation of D1 and D2 receptors is required for longterm synaptic depression in the striatum. Neurosci. Lett. 142, 9599. Charles A. et al. (2003) Coexpression of δopioid receptors with μreceptors in GH 3 cells changes the functional response to μagonists from inhibitory to excitatory. Mol. Pharm. 63 (1), 8995. Cheng Y.C. and Prusoff W.H. (1973) Relationship between the inhibition constant (K i) and the concentration of per cent inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 22, 30993108. Chio CL, Lajiness ME, Huff RM (1993). Activation of heterologously expressed D 3 dopamine receptors: comparison with D2 dopamine receptors. Am Soc Pharmacol Exp Ther 45: 5160. Chio C.L., Lajiness M.E. and Huff R.M. (1994) Activation of heterologously expressed D 3 dopamine receptors: comparison with D2 dopamine receptors. Mol. Pharmacol. 45, 5160. Cichon S., Nothen M.M., Stober G., Schroers R., Albus M., Maier W., Rietschel M., Korner J., Weigelt B., Franzek E., Wildenauer D., Fimmers R. and Propping P. (1996) Systenmatic screening for mutations in the 5’regulatory region of the human dopamine D1 receptor (DRD1) gene in patients with schizophrenia and bipolar affective disorder. Am. J. Med. Genet. 67,424 Civelli O., J.R. Bunzow, and D.K. Grandy (1993) Molecular diversity of the dopamine recptors. Annu. Rev. Pharmacol. 80 Toxicol. 32, 281307. Clementi C. e Fumagalli G. (1999) Farmacologia generale e cellulare. UTET Conklin B.R., Farfel Z., Lustig K.D., Julius D. and Bourne H.R. (1993) Substitution of three aminoacids switches receptor specificity of Gq to that of a Gi . Nature 363, 274276. Conklin B.R., Herzmark P., Ishida S., VoynoYasenetskaya T.A., Sun Y., Farfel Z. and Bourne H.R. (1996) Carboxylterminal mutation of Gq and Gs that alter the fidelity of receptor activation. Mol. Pharmacol. 50, 885890. Corsini G.U., Maggio R. and Vaglini F. (2002) Moleular and cellular events regulating dopamine neuron survival. Handbook of Exp.Pharm. 154 (II),321386 Crocq M.A., Mant R., Asherson P., Williams J., Hode Y., Mayerova A., Collier D., Lannfelt L., Sokoloff P., Schwartz J.C. (1992) Association between schizophrenia and Homozygsity at the dopamine D3 receptor gene. J. Med. Genet. 29, 858 Cullen B.R. (1987) Use of eukaryotic expression technology in the functional analysis of cloned genes. Methods Enzymol. 152, 684704. Cvejic S., and Devi L.A. (1997) Dimerization of the delta opioid receptor, implications for a function in receptor internalization. J. Biol. Chem. 272, 2695926964. Dal Toso R., Sommer B., Ewart M., Herb A., Pritchett D.B., Bach A., Shivers B.D. and Seeburg P.H. (1989) The dopamine receptor: two molecular forms generated by alternative splicing. EMBO J. 8, 40254034. De Camilli P., Macconi D. and Spada A. (1979) Dopamine inhibits adenylate cyclase in human prolactinsecreting pituitary adenomas. Nature 278, 252254. 81 Diaz L, Pilon C, Le Foll B, Gross C, Triller A, Schwartz JC et al (2000). Dopamine D3 receptors expressed by all mesencephalic dopamine neurons. J Neurosci 20: 86778684. Eason M.G., Kurose H., Holt B.D., Raymond J.R. and Ligget S.B. (1992) Simultaneous coupling of 2adrenergic receptors to two proteins with opposing effect: subtypes selective coupling of 2c2 adrenergic receptors to Gi and Gs. J. Biol. Chem. 267, 1579515801. Eason M.G. and Ligget S.B. (1995) Identification of a Gs coupling domain in the amino terminus of the third intracellular loop of the 2A adrenergic receptor. J. Biol. Chem. 270, 2475324760. Enjalbert A. and Bockaert J. (1983) Pharmacological characterization of the D 2 dopamine receptor negatively coupled with adenylate cyclase in rat anterior pituitary. Mol. Pharmacol. 53, 576584. Filteau F., Veilleux F. and Lévesque D. (1999) Effects of reciprocal chimeras between the Cterminal portion of third intracellular loops of the human dopamine D2 and D3 receptors. FEBS Lett. 447, 251256. Fishburn C.S., Belleli D., David C., Carmon S. and Fuchs S. (1993) A novel short isoform of the D3 dopamine receptor generated by alternative splicing in the third cytoplasmic loop. J. Biol. Chem. 268, 58725878. Florio V.A. and Sternweiss P.C. (1985) Reconstitution of resolved muscarinic cholinergic receptors with purified GTP binding proteins. J. Biol. Chem. 260, 34773483. Freedman S.B., Patel S., Marwood R., Emms F., Seabrook G.R., Knowles M.R. and McAllister G. (1994) Expression and pharmacological characterization of the human D3 dopamine receptor. J. Pharmacol. Exp. Ther. 268, 417426. Fukushima Y., Asano T., Saitoh T., Anai M., Funaki M., Ogihara T., Katagigi H., Matsuhashi N., Yazaki Y. and Sugano K. (1997) Oligomer formation of histamine H2 receptors expressed in Sf9 and COS7 cells. FEBS Lett. 409, 283286. Galvez T., Duthey B., Kniazeff J., Blahos J., Rovelli G., Bettler B., Prézeau L. and Pin J. (2001) Allosteric interactions between GB1 and GB2 subunits are required for optimal GABAb receptor function. EMBO J. 20 (9), 21522159. 82 Ganz M.B., Patcher J.A., and Barber D.L. (1990) Multiple receptors coupled to adenylyl cyclase regulate Na+/H+ exchange indipendent of cAMP. J. Biol. Chem. 265, 89898992. Gelernter J. et al. (1992) the D4 dopamine receptor (DRD4) maps to distal 11p close to HRAS. Genomics 13, 208. George S.R., Lee S.P., Varghese G., Zeman P.R., Seeman P., Ng G.Y.K. and O’Dowd B.F. (1998) A transmembrane domainderived peptide inhibits D1 dopamine receptor function without affecting receptor oligomerization. J. Biol. Chem. 46, 3024430248. Gingrich J.A., and Caron M.G. (1993) Recent advances in the molecular biology of dopamine receptors. Annu. Rev. Neurosci. 16, 299321. Giros B., Sokoloff P., Martres M.P., Riou J.F., Emorine L.J. and Schwartz J.C. (1989) Alternative splicing directs the expression of two D2 dopamine receptor isoforms. Nature 342, 923926. Glatt C.E. and Snyder S.H. (1993) Cloning and expression of an adenylyl cyclase localized to the corpus striatum. Nature (Lond.) 361, 536538. Gobert A, Rivet JM, Cussac D, NewmanTancredi A, Lejeune F, Bosc C et al (2000). S33592, a benzopyranopyrrole partial agonist at dopamine D2/D3 receptors and potential antipsychotic agent: I. Modulation of dopaminergic transmission in comparison to aripiprazole, preclamol and raclopride. Am Soc Neurosci 26: 274. Gouldson P.R. and Reynolds C.A. (1997) Simulation on dimeric peptides evidence for domain swapping in Gprotein coupled receptors. Biochem. Soc. Trans. 25, 10661071. Gouldson P.R., Snell C.R. and Reynolds C.A. (1997) A new approach to docking in the 2adrenergic receptor which exploits the domain structure of Gprotein coupled receptors. J. Med. Chem. 40, 38713886. 83 Gouldson P.R., Snell C.R., Bywater R.P., Higgs C. and Reynolds C.A. (1998) Domain swapping in Gprotein coupled receptor dimers. Protein eng. 11, 11811193. Grandy D.K., Litt M., Allen L., Bunzow J.R., Marchionni M., Makam H., Reed L., Magenis R.E. and Civelli O. (1989) The human dopamine D2 receptor gene is located on chromosome 11 at q22q23 and identifies a taqI RFLP. Am. J. Hum. Genet. 45, 778. Grosse R., Schöneberg T., Schultz G. and Gudermann T. (1997) Inhibition of gonadotropinrealising hormone receptor signaling by expression of a splice variant of the receptor. Mol. Endocrinol. 11 (9), 13051318. Gurevich EV, Joyce JN (1999). Distribution of dopamine D3 receptor expressing neurons in the human forebrain: comparison with D2 receptor expressing neurons. Neuropsychopharmacology 20: 6080. Havlickova M., Prézeau L., Duthey B., Bettler B., Pin J. and Blahos J. (2002) The intracellular loops of the GB2 subunit are crucial for Gprotein coupling of the hetrromeric γaminobutyrate B receptor. Mol. Pharm. 62, 343350. Hulme E.C., Birdsall N.J.M. and Buckley N.J. (1990) Muscarinic receptor subtypes. Annu. Rev. Pharmacol. Toxicol. 30, 633673. Jensen A.A., Pederson U.B., Kiemer A., Din N. and Andersen P.H. (1995) Functional importance of the carboxyl tail cysteine residues in the human D1 dopamine receptor. J. Neurochem. 65, 13251331. Jones K.A., Boronsky B., Tamm J.A., Craig D.A., Durkin M.M., Dai M., Yao W.J., Jonson M., Gunwaldsen C., Huang L.Y., Tang C., Shen Q., Salon J.A., Morse K., Laz T., Smith K.E., Nagarathnam D., Noble S.A., Branchek T.A. and Gerald C. (1998) GABAB receptors function as a heterotrimeric assembly of the subunits GABA BR1 and GABABR2. Nature 396, 674 679. Jordan M., Schallhorn A., Wurm F.M. (1996)Transfecting mammalian cells: optimization of critical parameters affecting calciumphosphate precipitate formation. Nucleic acids research24 ,596601. 84 Jordan B.A. and Devi L.A. (1999) Gproteins coupled receptor heterodimerization modulates receptor function. Nature 399, 697700. Jordan B.A., Cvejic S. and Devi L.A. (2000) Opioids and their complicated receptor complexes. Neuropsychopharmacology 23, NO.S4. Jordan B.A., Trapaidze N., Gomes I., Nivarthi R. and Devi L. (2001) Oligomerzation of opioid receptors with β 2 adrenergic receptors: a role in trafficking and mitogenactived protein kinase activation. PNAS 98, 343348. Joyce JN (2001). Dopamine D3 receptors as a therapeutic target for antipsychotic and antiparkinsonian drugs. Pharmacol Ther 90: 231259. Karpa K.D., Lin R., Kabbani N. and Levenson R. (2000) The dopamine D 3 receptor interacts with itself and the truncated D3 splice variant D3nf: D3D3nf interaction causes mislocalization of D3 receptors. Mol. Pharmacol. 58, 677683. Kebabian J.W. and Calne D.B. (1979) Multiple receptors for dopamine. Nature 277, 9396. Keefe K.A. and Gerfen C.R. (1995) D1D2 dopamine receptor synergy in striatum: effects of intrastriatal infusion of dopamine agonist and antagonist on immediate early gene expression. Neuroscience 66, 903913. Kelly M.A., Rubinstein M., Asa S.L., Zhang G., Saez C., Bunzow J.R., Allen R.G., Hnasko R., BenJonathan N., Grandy D.K. and Low M.J. (1997) Pituitary lactotroph hyperplasia and chronic hyperprolactinemia in dopamine D2 receptor deficient mice. Neuron 19, 103113. Kobilka B.K., Kobilka T.S., Daniel K., Regan J.W., Caron M.G. and Lefkowitz R.J. (1988) Chimeric 2, 2adrenergic receptors: delineation of domains involved in effector coupling and ligand binding specificity. Science 240, 13101316. Kojima H., Ohmori O., Shinkai T., Terao T., Suzuki T. and Abe H. (1999) Dopamine D 1 receptor gene polimorphism and 85 schizophrenia in Japan. Am. J. Med. Genet. 88, 116. Kozell L.B., Macida C.A., Neve R.L. and Neve K.A. (1994) Chimeric D1/D2 dopamine receptors. Distinct determinants of selective efficacy, potency and signal trasduction. J. Biol. Chem. 269, 3029930306. Kunishima N. et al. (2000) Structural basis of glutamate recognition by dimeric metabotropic glutamate receptors. Nature 407, 971. Lachowicz J.E. and Sibley D.R. (1997) Chimeric D2/D3 dopamine receptor coupling to adenylyl cyclase. Biochem. Biophys. Res. Commun. 237, 394399. Landwehrmeyer B., Mengod G. and Palacios J.M. (1992) Dopamine D3 receptor mRNA and binding sites in human brain. Mol. Brain Res. 18, 187192. Lawler CP, Prioleau C, Lewis MM, Mak C, Jiang D, Schetz JA et al (1999). Interactions of the novel antipsychotic aripiprazole (OPC14597) with dopamine and serotonin receptor subtypes. Neuropsychopharmacology 20: 612627. Lerer B. et al. (2002) Pharmacogenetics of tardive dyskinesia: combined analysis of 780 patients supports association with dopamine D3 receptor gene Ser9Gly polymorphism. Neuropsychopharmacology 27 (1), 105. Levesque D., Diaz J., Pilon C., Matres M., Giros B., Souil E., Schott D., Morgat J., Schwartz J., and Sokoloff P. (1992) Identification, characterization, and localization of the dopamine D3 receptor in rat brain using 7[3H]hydroxyN,Ndin Propyl2Aminotetrail. PNAS 89, 81558159. Li Z, Ichikawa J, Dai J, Meltzer HY (2004). Aripiprazole, a novel antipsychotic drug, preferentially increases dopamine release in the prefrontal cortex and hippocampus in rat brain. Eur J Pharmacol 493: 7583. 86 Lichter J.B., Barr C.L., Kennedy J.L, Van Tol H.H.M., Kid K.K. and Livak K.J. (1993) A hypervariable segment in the human dopamine D4 (DRD4) gene. Hum. Mol. Genet. 2, 767773. Liu K., Bergson C., Levenson R. and Schmauss C. (1994) On the origin of mRNA encoding the truncated dopamine D3 type receptor D3nf and detection of D3nflike immunoreactivity in human brain. Yric J. Biol. Chem. 269, 2922029226. Liu K. et al. (2000) Direct proteinprotein coupling enables crosstalk between dopamine D5 and γaminobutyric acid A receptor. Nature 403, 274. Lowry O.H., Rosebrough N.J., Farr A.L. and Randhall R.J. (1951) Protein measurement with the Folin Phenol reagent. J. Biol. Chem. 193, 265275. Maggio R., Vogel Z. and Wess J (1993a) Coexpression studies with mutant muscarinic/adrenergic receptors provide evidence for intermolecular “crosstalk” between Gproteinlinked receptors. Proc. Natl. Acad. Sci. USA 90, 31033107. Maggio R., Vogel Z. and Wess J. (1993b) Reconstitution of functional muscarinic receptor by coexpression of amino and carboxylterminal receptor fragments. FEBS Lett. 319, 195200. Maggio R., Barbier P., Colelli A., Salvadori F., DeMontis G. and Corsini G.U. (1999) Gproteinlinked receptors: pharmacological evidence for the formation of heterodimers. J. Pharmacol. Exp. Ther. 291, 251257. Maggio R, Scarselli M, Novi F, Millan MJ, Corsini GU (2003). Potent activation of dopamine D3. D2 heterodimers by the antiparkinsonian agents, S 32504, pramipexole and ropinirole. J Neurochem 87: 631641. Maggio R, Novi F, Scarselli M, Corsini GU (2005). The impact of Gproteincoupled receptor heterooligomerization on function and pharmacology. FEBS J 272: 29362946. Marek K. et al. (2002) Dopamine trasporter brain imaging to assess the effects of pramipexole vs Levodopa on Parkinson Disease Progression. JAMA 287 (13), 1653. 87 McDonald W.M., Sibley D.R., Kilpatrick B.F. and Caron M.G. (1984) Dopaminergic inhibition of adenylate cyclase correlates with high affinity agonist binding to anterior pituitary D2 dopamine receptors. Mol. Cell. Endocrinol. 36, 201209. MeadorWoodruff JH, Damask SP, Wang J, Haroutunian V, Davis KL, Watson SJ (1996). Dopamine receptor mRNA expression in human striatum and neocortex. Neuropsychopharmacology 15: 1729. Monnot C., Bihoreau C., Conchon S., Curnow K.M., Corvol P. and Klauser E. (1996) Polar residues in the transmembrane domains of the type 1 angiotensin II receptor are required for binding and coupling. Reconstitution of the binding site by coexpression of two deficient mutants. J. Biol. Chem. 271, 15071513. Mons N. and Cooper D.M.F. (1994) Selective expression of one Ca ++inhibitable adenylyl cyclase in dopaminergically innervated rat brain regions. Mol. Brain Res. 22, 236244. Monsma F.J. Jr, McVittie L.D., Gerfen C.R., Mahan L.C., Sibley D.R., Richardson R.M. and Hosey M.M. (1989) Multiple D 2 dopamine receptors produced by alternative RNA splicing. Nature 342,926929. Morissette M, Goulet M, Grondin R, Blanchet P, Bedard PJ, Di Paolo T et al (1998). Associative and limbic regions of monkey striatum express high levels of dopamine D3 receptors: effects of MPTP and dopamine agonist replacement therapies. Eur J Neurosci 10: 25652573. Ng G.Y.K., Mouillac B., George S.R., Caron M., Dennis M., Bouvier M. and O’Dowd B.F. (1994) Desensitization, phosphorilation and palmitoylation of the human dopamine D1 receptor. Eur. J. Pharmacol. (Mol. Pharmacol. Sect.) 267, 7 19. Nielsen S.M., Elling C.E. and Schwartz T.W. (1998) Splitreceptors in the tachykinin neurokinin1 systemmutational analysis of intracellular loop 3. Eur. J. Biochem. 251, 217. 88 Nimchinsky E.A., Hof P.R., Janssen W.G.M., Morrison J.H. and Schmauss C. (1997) Expression of dopamine D 3 receptor dimers and tetramers in brain and in transfected cells. J. Biol. Chem. 272, 2922929237. Obadiah J., AvidorReiss T., Fishburn C.S., Carmon S., Bayewittch M., Vogel Z., Fuchs S. and LevaviSivan B. (1999) Adenylyl cyclase interaction with the D2 dopamine receptor family: differential coupling to Gi, Gz and Gs. Cell. Mol. Neurobiol. 19, 653664. O’Dowd B.F. (1993) Structure of dopamine receptors. J. Neurochem. 60, 804816. Oliveri R.L., Annesi G., Zappia M., Civitelli D., Montesanti R., Branca D., Nicoletti G., Spadafora P., Pasqua A.A., Cittadella R., Andreoli V., Gambardella A., Aguglia U. and Quattrone A. (1999) Dopamine D2 receptor gene polymorphism and the risk of levodopainduced dyskinesias in PD. Neurology 53,1425. Onali P.L., Schwartz J.P. and Costa E. (1981) Dopaminergic modulation of adenylate cyclase stimulation by vasoactive intestinal peptide (VIP) in anterior pituitary. Proc. Natl. Acad. Sci. USA 78, 65316534. Onali P.L., Olianas M.C. and Gessa G.L. (1985) Characterization of dopamine receptors mediating inhibition of adenylate cyclase activity in rat striatum. Mol. Pharmacol. 28: 138145. Palczewski K. et al. (2000) Crystal structure of rhodopsin: a G proteincoupled receptor. Science 28, 739745. Park PS, Filipek S, Wells JW, Palczewski K (2004). Oligomerization of G proteincoupled receptors: past, present, and future. Biochemistry 43: 1564315656. Pfeiffer M. et al. (2002) Heterodimerization of somatostatin and opioid receptors crossmodulates phosphorylation, internalization and desensitation. J.Biol.Cem. 277 (22), 1976219772. Pilon C., Levesque D., Dimitriadou V., Griffon N., Martres M.P., Schwartz J.C. and Sokoloff P. (1994) Functional coupling of the human dopamine D3 receptor in a transfected NG 10815 nueroblastomaglioma hybrid cell line. Eur. J. Pharmacol. 268, 129139. 89 Piomelli D., Pilon B., Giros B., Sokoloff P., Matres M.P. and Schwartz J.C. (1991) Dopamine activation of the arachidonic acid cascade as basis for D1/D2 receptor synergism. Nature 353, 164164. Pontieri F. and Di Chiara G. (1995) Intravenous cocaina, morphine and amphetamine preferentially increase extracellular dopamin in the “schell” as compared with “core” of the rat nucleus accumbens. PNAS 92, 1230412308. Potenza M.N., Graminski G.F., Schmauss C. and Lerner M.R. (1994) Functional expression and characterization of human D2 and D3 dopamine receptors. J. Neurosci. 14, 14631476. Quik M, Police S, He L, Di Monte DA, Langston JW (2000). Expression of D(3) receptor messenger RNA and binding sites in monkey striatum and substantia nigra after nigrostriatal degeneration: effect of Levodopa treatment. Neuroscience 98: 263273. Richardson R.M. and Hosey M.M. (1992) Agonistinduced phosphorylation and desensitization of human M2 muscarinic cholinergic receptors in Sf9 insect cells. J. Biol. Chem. 267, 2224922255. Ridge K.D., Lee S.S.J. and Yao L.L. (1995) In vivo assembly of rhodopsin from expressed polypeptide fragments. Procl. Natl. Acad. Sci. USA 92, 32043208. Rivet JM, Brocco M, Dekeyne A, Dubuffet T, Lavielle G, Millan MJ (2000). S33592; a benzopyranopyrrole partial agonist at dopamine D2/D3 receptors and potential antipsychotic agent: II. Functional profile in comparison to aripiprazole, preclamol and raclopride. Am Soc Neurosci 26: 272. Robinson SW, Caron MG (1997). Selective inhibition of adenylyl cyclase Type V by the Dopamine D 3 receptor. Mol Pharmacol 52: 508514. Romano C., Yang W.L. and O’Malley K.L. (1996) Metabotropic glutamate receptor 5 is a disulfidelinked dimer. J. Biol. 90 Chem. 271, 2861228616. Saiardi A., Bozzi Y., Baik J.H. and Borrelli E. (1997) Antiproliferative role of dopamine loss of D2 receptors causes hormonal dysfunction and pituitary hyperplasia. Neuron 19, 115126. Scarselli M., Armogida M., Chiacchio S., DeMontis M.G., Colzi A., Corsini G.U. and Maggio R. (2000) Reconstitution of functional dopamine D2s receptor by coexpression of amino and carboxylterminal receptor fragments. Eur. J. Pharmacol. 397, 291296. Scarselli M, Novi F, Schallmach E, Lin R, Baragli A, Colzi A et al (2001). D2/D3 dopamine receptor heterodimers exhibit unique functional properties. J Biol Chem 276: 3030830314. Shapiro DA, Renock S, Arrington E, Chiodo LA, Liu LX, Sibley DR et al (2003). Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology 28: 14001411. Schinelli S., Paolillo M. and Corona G.L. (1994) Opposing actions of D 1 and D2 dopamine receptors on arachidonic acid release and cyclic AMP production in striatal neurons. J. Neurochem. 62, 944949. Schöneberg T., Liu J. and Wess J. (1995) Plasma membrane localization and functional rescue of truncated forms of G protein coupled receptor. J. Biol. Chem. 270, 1800018006. Schöneberg T., Yun J., Wenkert D. and Wess J. (1996) Functional rescue of mutant V 2 vasopressin receptors causing nephrogenic diabetes insipidus by a coexpressed receptor polypeptide. EMBO J. 15, 12831291. Schulz A., Grosse R., Schultz G., Gudermann T. and Schöneberg T. (1999) Structural implication for receptor oligomerization from functional reconstitution studies of mutant V2 vasopressin receptors. J. Biol. Chem. 275, 23812389. 91 Seabrook G.R., Kemp J.A., Freedman S.B., Patel S., Sinclair H.A. and McAllister G. (1994) Functional expression of human D3 dopamine receptors in differentiated neuroblastoma x glioma NG10815 cells. Br. J. Pharmacol. 111, 391393. Seeman P. and Van Tol H.H.M. (1994) Dopamine receptor pharmacology. TIPS Rev. 15, 264270. Snyder L.A., Roberts J. and Sealfon S. (1991) Alternative trascripts of the rat and human dopamine D3 receptor. Biochem. Biophys. Res. Commun. 180, 10311035. Segal DM, Moraes CT, Mash DC (1997). Upregulation of D3 dopamine receptor mRNA in the nucleus accumbens of human cocaine fatalities. Brain Res Mol 45: 335339. Sokoloff P., Giros B., Martres M.P., Bouthenet M.L. and Schwartz J.C. (1990) Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 347, 146151. Sokoloff P., Andrieux M., Besancon R., Pilon C., Martres M.P., Giros B. (1992 ) Pharmacology of Human dopamine D3 receptor expressed in mammalian cell line : comparison with D2 receptor. European Jurnal of Pharmacology – Molecular Pharmacology section 225, 331337. Spano P.F., Govoni S. and Trabucchi M. (1978) Studies on the pharmacological properties of dopamine receptors in various areas of the central nervous system. Adv. Biochem. Psychopharmacol. 19, 155165. Spiegel A.M. (1996) Defects in Gproteincoupled signal transduction in human desease. Annu. Rev. Physiol. 58, 143170. Staley JK, Mash DC (1996). Adaptive increase in D3 dopamine receptors in the brain reward circuits of human cocaine fatalities. J Neurosci 16: 61006106. Stanwood GD, Artymyshyn RP, Ki_ng MP, Kung HF, Lucki I, McGonigle P (2000). Quantitative autoradiographic mapping of rat brain dopamine D3 binding with [125I]7OHPIPAT: evidence for the presence of D3 receptors on dopaminergic and nondopaminergic cell biodies and terminals. J Pharmacol Exp Ther 295: 12231231. 92 Sunahara R.K., Guan H.C., O’Dowd B.F., Seeman P., Laurier L.G., George S.R., Torchia J., Van Tol H.H.M. and Niznik H. (1991) Cloning a human dopamine receptor gene (D5) with higher affinity for dopamine than D1. Nature 350, 614619. Sunahara R.K., Dessauer C.W. and Gilman A.G. (1996) Complexity and diversity of mammalian adenylyl cyclase. Annu. Rev. Pharmacol. Toxicol. 36, 461480. Surmeier D.J., WenJie S. and Zhen Y. (1996) Coordinated expression of dopamine receptors in neostriatal medium spiny neurons. J. Neurosci. 16, 65796591. Suzuki M., Hurd Y.L., Sokoloff P., Schwartz J.C. and Sedvall G. (1997) D 3 dopamine receptor mRNA is widely expressed in the human brain. Brain Res. 779, 5874. Swarzenski B., Tang Y.J., Oh Y., O'Malley K.L. and Todd R.D. (1994) Morphogenic potentials of D2, D3, D4 dopamine receptors revealed in trasfected neuronal cell lines. PNAS 91, 649653. Tadori Y, Miwa T, Tottori K, Burris KD, Stark A, Mori T et al (2005). Aripiprazole’s low intrinsic activities at human dopamine D2L and D2S receptors rendrer it a unique antipsychotic. Eur J Pharmacol 515: 1019. Tang L., Todd R.D., O'Malley K.L. (1994) Dopamine D2 and D3 receptors inhibit dopamine release. J.Pharm.Exp.Ther. 268, 495502. Urlaub G., Chasin L.A., (1980) Isolation of Chinese hamster cell mutants deficient in dihydrofolate reductase activity. Proc.Natl.Acad.Sci.USA 77, 42164220. Vallar L., Muca C.M., Albert P., Bunzow J., Meldolesi J. and Livelli O. (1990) Differential coupling of dopaminergic D 2 receptors expressed in different cell types: stimulation of PI 4,5 biphosphate hydrolisis in LTK fibroblasts hyperpolarization and cytosolicfree Ca++ concentration decrease in GH4 C1 cells. J. Biol. Chem. 265, 1032010326. 93 Vanhauwe J., Freyman N., Francken J., Walter H., Luyten M. and Leysen J. (1999) Comparision of the ligand binding and signaling properties of Humane dopamine D2 and D3 receptor in Chinese hamster ovary cells. J.Pharm.Exp.Ther. 290, 908916. Van Tol H.H.M., Bunzow J.R., Guan H.G., Sunhara R.K., Seeman P., Niznik H. and Civelli O. (1991) Cloning of the gene for a human dopamine D4 receptor with high affinity for the antipsychotic clozapine. Nature 350, 610614. Van Tol H.H.M., Wu C.M., Guan H.C., Ohara K., Bunzow J.R., Civelli O., Kennedy J., Seeman P., Niznik H. and Jovanovic V. (1992) Multiple dopamine D4 receptor variants in the human population. Nature 358, 149152. Van Vliet BJ, Mos J, Van der Heijden JAM, Feenstra R, Kruse CG, Long SK (2000). DU 127090: a highly potent, atypical dopamine receptor ligand a putative potent full spectrum antipsychotic with low EPS potential. Eur Neuropsychopharmacol 10: P.2.034. Wang J., Zhuo L. and Chen B. (2001) Association study of dopamine D2, D3 receptors gene polimorphism with motor fluctuations in PD. Neurology 56, 17571759. Wess J., Liu J., Blin N., Yun J., Lerche C. and Kostenis E. (1997) Structural basis of receptor/G protein coupling selectivity studied with muscarinic receptor as a model systems. Life Sci. 60, 10071014. Wong A.H.C., Buckle C.E. and Van Tol H.H.M. (2000) polimorphism dopamine receptors.What do they tell us ? Eur. J. Pharm 410, 183203. Xie Z., Lee S.P., O’Dowd B.F. and George S.R. (1999) Serotonin 5HT1B and 5HT1D receptors form homodimers when expressed alone and heterodimers when coexpressed. FEBS Lett. 456, 6367. 94 Zawarinsky P., Tallerico T., Seeman V., Lee S.P., O’Dowd B.F. and George S.R. (1998) Dopamine D2 receptor dimers in human and rat brain. FEBS Lett. 441, 383386. Zhang L.J., Lachowicz J.E. and Sibley D.R. (1994) The D2s and D2l dopamine receptor isoforms are differentially regulated in Chinese Hamster Ovary cells. Mol. Pharmacol. 45, 878889. Zhu C. C., Cook L. B. And Hinkle P.M. (2002) Dimeritation and phosphorilation of thyrotropinrelesing hormone receptors are modulated by agonist stimulation. J. Biol. Chem. 277, 2822828237. 95 ABBREVIAZIONI AC V/VI adenilato ciclasi chimerica V/VI AMP adenosina monofosfato cAMP AMP ciclico 3 HcAMP AMP ciclico triziato CHO ovario di criceto cinese COS7 rene di scimmia verde africana DA dopamina DAG diacilglicerolo DEAE dietilaminoetil DMEM medium di Eagle modificato da Dulbecco DHFR deidrofolato reduttasi DNA acido desossiribonucleico EDTA agente chelante il ferro ERKs chinasi regolate da segnali extracellulari FBS siero fetale bovino GABA acido γamminobutirrico GPCRs recettori accoppiati alle proteine G GTP, GDP, GMP guanosina trifosfato, guanosina difosfato, guanosina monofosfato IP3 inositolo trifosfato i3 loop terzo loop intracellulare MAPKs proteine chinasi attivate da mitogeni NaCl cloruro di sodio NDmc NDesmetilclozapina PBS tampone fosfato salino PIP2 fosfatidilinositolo bifosfato PKA proteina chinasi A PKC proteina chinasi C PLC fosfolipasi C SNA sistema nervoso autonomo TM transmembrana 96 97
Scaricare