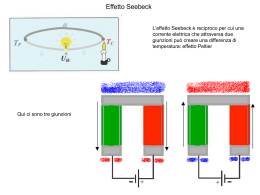
La Struttura dei Cristalli
Distanza fra atomi/molecole
Densità
Solido
pochi Å
Gassoso
~ 30 Å
Liquido
fra i due precedenti
1022 ÷1023 atomi/cc
1019 molecole/cc (T, P
standard)
fra i due precedenti
SOLIDI. Si suddividono in solidi cristallini e solidi amorfi.
Solidi cristallini : atomi sono disposti in modo regolare con struttura tridimensionale
ordinata con ripetizione periodica e regolare di una stessa unità di base. La più piccola unità
ripetente in un cristallo è detta “cella”, la disposizione spaziale periodica della cella è detta
“reticolo”.
• Monocristalli: all’interno di essi gli atomi sono disposti regolarmente per tutta
l’estensione del pezzo.
• Policristalli: insieme di singoli cristalli uniti tra loro, detti“grani”. L’orientazione dei diversi
grani l’uno rispetto all’altro è solitamente casuale.
Solidi Amorfi: In un solido amorfo gli atomi sono disposti casualmente. Si ha una certa
regolarità nella disposizione atomica solo su scale di ridottissime dimensioni.
b
c
STRUTTURE CRISTALLINE
La cella elementare è la più piccola unità
ripetente che mostra pienamente le
simmetrie e la struttura del cristallo.
d
a
La cella elementare di base in tre dimensioni è
un parallelepipedo.
La lunghezza di ogni lato è dato da a, b e c
(parametri della cella elementare). L’angolo fra i
lati è dato dagli angoli a, b e g. Il regolare
impilamento di questi parallelepipedi da origine
ad una struttura tridimensionale
Esempio: NaCl.
Gli ioni Na+ sono ai vertici e ai centri delle facce,
gli ioni Cl- sono al centro degli spigoli della cella
cubica.
SETTE SISTEMI CRISTALLINI
Reticoli di Bravais
Dal momento che i punti reticolari
della cella elementare possono essere
sistemati in modi diversi, all’interno di
ogni sistema cristallino sono possibili
diversi tipi di reticolo.
Ci sono 14 diversi modi di distribuire
punti reticolari per formare reticoli
spaziali. Questi 14 reticoli sono detti
reticoli di Bravais.
INDICIZZAZIONE DELLE DIREZIONI E DEI PIANI CRISTALLOGRAFICI.
INDICI DI MILLER E DI MILLER-BRAVAIS
Si impiega un riferimento di assi ortogonali x, y, z con la convenzione che l’asse x ha come
Verso positivo quello uscente dal foglio
DIREZIONI. Si può seguire la seguente procedura:
1- determinare le coordinate dei punti che giacciono lungo la direzione di interesse;
2 – sottrarre alle coordinate della ‘punta’ le coordinate della ‘coda’;
3 – eliminare le frazioni e/o ridurre i risultati ottenuti ai minimi interi;
4 – racchiudere gli indici entro parentesi quadra. Se sono presenti segni negativi,
tracciare una barretta su quel numero
NOTA.
• Una direzione e il suo negativo non sono identici: [100] è diverso da [-100]
• Una direzione e un suo multiplo sono direzioni identiche: [100] = [200] = [300]
• Certi gruppi di direzioni sono equivalenti, in quanto hanno degli indici particolari
solo per come si è posizionato il riferimento. Ad es.:[110] = [101] = [011] = [-110] =
= [1-10] = [10-1] = [-10-1] = [0-11]; un gruppo di direzioni equivalenti si indica
adottando la notazione <110>
PIANI. Si può seguire la seguente procedura:
1- identificare le intercette del piano con gli assi x, y, z in termini di
numero di parametri cristallini;
2 – fare il reciproco delle intercette;
3 – eliminare le frazioni senza ridurre agli interi più piccoli;
4 – racchiudere gli indici entro parentesi tonda. Se sono presenti segni
negativi, tracciare una barretta su quel numero
NOTA.
•Un piano e il suo negativo non sono identici: (020) è diverso da (0-20)
•Un piano e i suoi multipli non sono identici, poiché possono differire i
valori di densità planare e frazione di impacchettamento planare;
•Certi gruppi di piani sono equivalenti, in quanto hanno degli indici
particolari solo per come si è posizionato il riferimento. Ad es.:(110) =
(101) = (011) = (-110) = (1-10) = (10-1) = (-10-1) = (0-11); un gruppo di
direzioni equivalenti si indica adottando la notazione {110}
SISTEMA ESAGONALE – INDICI DI MILLER BRAVAIS A 4 ASSI (a1,a2,a3,c)
DIREZIONI.
Si determina il numero di parametri cristallini necessari per spostarsi dalla ‘coda’
Alla ‘punta’ del vettore indicante la direzione.
PIANI.
1- identificare le intercette del piano con gli assi x, y, z in termini di numero di parametri
cristallini;
2 – fare il reciproco delle intercette;
3 – eliminare le frazioni senza ridurre agli interi più piccoli;
4 – racchiudere gli indici entro parentesi tonda. Se sono presenti segni negativi, tracciare
una barretta su quel numero
NOTA. Con il sistema a 4 assi si ha sempre (hkil) con
h + k = -i
STRUTTURE CRISTALLINE
METALLICHE
I metalli sono caratterizzati da elevato
numero di atomi primi vicini e da strutture
compatte. Se si rappresentano gli atomi
come sfere rigide di raggio pari al raggio
atomico, queste hanno tipicamente
dimensioni di 0.10.2 nm. I più comuni tipi
di celle elementari dei metalli sono tre:
• FCC (faced centered cubic): Cubico a
facce centrate;
• BCC (body centered cubic): Cubico a
corpo centrato;
• HCP (hexagonal close-packed): Esagonale
compatto.
“numero di coordinazione” (CN, coordination number) : numero di atomi con i
quali un atomo è legato, cioè il numero di sfere tangenti una data sfera.
“numero di atomi per cella elementare”:valore unitario intero ad ogni
atomo appartenente esclusivamente alla cella data, ½ ad ogni atomo posizionato al
centro di una faccia, ¼ a quelli posizionati su uno spigolo e 1/8 a quelli sui vertici.
“fattore d’impacchettamento atomico” (APF, atomic packing factor) :
frazione di volume occupata dalle sfere all’interno della cella.
STRUTTURE CRISTALLINE METALLICHE
Numero di coordinazione
è il numero di atomi con i quali un atomo è legato,
cioè il numero di sfere tangenti una data sfera.
FCC:
BCC:
HCP:
12
8
12
Numero atomi per cella elementare
Numero di atomi “interi” contenuti all’interno della cella elementare
- Gli atomi che appartengono a due celle diverse (cioè si trovano al centro
delle facce) contano per ½.
- Gli atomi che appartengono a 4 celle diverse (centro degli spigoli in cella
cubica) contano per ¼.
- Gli atomi che si trovano sui vertici della cella (cubica) contano per ⅛.
FCC: 4
BCC: 2
HCP: 6
STRUTTURE CRISTALLINE METALLICHE
Struttura cubica a corpo centrato
Esempi: Cr, Mo, a-Fe …
NUMERO DI COORDINAZIONE = 8
ATOMI/CELLA =2
STRUTTURE CRISTALLINE METALLICHE
Struttura cubica a facce centrate
Esempi: Cu, Al, Ag, Au, ...
NUMERO DI COORDINAZIONE = 12
ATOMI/CELLA = 4
STRUTTURE CRISTALLINE METALLICHE
Struttura esagonale compatta
Esempi: Cd, Mg, Zn, Ti, …
NUMERO DI COORDINAZIONE = 12
ATOMI/CELLA =6
STRUTTURE CRISTALLINE METALLICHE
Approssimazione di sfere rigide:
consideriamo gli atomi come sfere perfettamente
rigide poste nelle posizioni di un reticolo cristallino
a contatto fra loro.
Date le dimensioni del raggio atomico possiamo dedurre le dimensioni della cella elementare
FCC:
le sfere si toccano lungo la diagonale di
una faccia.
Fra le dimensioni atomiche e quelle della cella elementare
vale la relazione:
2 a 4 R
BCC:
le sfere si toccano lungo la diagonale
della cella
Fra le dimensioni atomiche e quelle della cella elementare
vale la relazione:
3 a 4 R
STRUTTURE CRISTALLINE METALLICHE
Calcolo del fattore d’impacchettamento atomico
Frazione della cella elementare occupata dagli atomi =
numero di atomi per cella elementare volume atomico
volume cella elementare
2 a 4 R
FCC:
a 2 2 R
Vcella a 3 16 2 R 3
4
16
Voccupato 4 R 3 R 3
3
3
APF
Voccupato
Vcella
16
16
R3
R3
3 3
3
0,74
3
a
16 2 R
3 2
3 a 4 R
BCC:
a
4
R
3
Vcella a 3
4
8
Voccupato 2 R 3 R 3
3
3
APF
Voccupato
Vcella
8
8
R3
R3
3
3 3
3
0,68
64
a
8
R3
3 3
64
3 3
R3
Densità Planari e Lineari
Il concetto di equivalenza di piani e direzioni cristallografiche riflette il fatto che piani e
direzioni equivalenti presentano rispettivamente densità planari e lineari uguali. Il
valore di densità planari e lineari è fondamentale per comprendere le proprietà di
deformazione plastica di un materiale metallico.
Calcoliamo la densità planare deI pianI 110 di un reticolo FCC.
(110)
2 a 4 R
a 2 2 R
Atotale a 2 a (2 2 R) 2 (2 2 R) 8 2 R 2
1
1
Aoccupata 2 R 2 4 R 2 2 R 2
2
4
d planare(110) FCC
2 R2
0,555
2
8 2 R
4 2
NOTA: CON LO STESSO METODO SI CALCOLA LA DENSITA’ PLANARE DEI PIANI DI MASSIMO
ADDENSAMENTO ATOMICO 111 PER CUI RISULTA d (111) FCC=
0,9
Densità lineare della direzione <100> di un reticolo
BCC.
La lunghezza occupata è pari a:
2 R
z
La relazione tra R e a nel c.c.c. è già stata trovata:
3 a 4 R
a
4
R
3
y
[100]
x
la densità lineare lungo <100> sarà:
d lineare[100]
3
2 R
0,866
4
2
R
3
NOTA: NATURALMENTE CON LO STESSO METODO SI CALCOLA LA DENSITA’ LINEARE
DELLE DIREZIONI DI MASSIMO ADDENSAMENTO ATOMICO <111> PER CUI RISULTA
d <111> FCC= 1
CALCOLO DENSITA’ TEORICA
r
La densità di un materiale cristallino, , sarà pari alla densità della cella
elementare,cioè pari al rapporto fra il numero di atomi nella cella per la massa di un
atomo e il volume della cella:
n A
r
Vc N A
n = numero di atomi nella cella elementare (BCC=2, FCC=4, HCP=6);
A = peso atomico;
Vc = volume della cella elementare
NA = numero di Avogadro (pari al numero di atomi in una mole)
Peso atomico e raggio atomico si possono trovare sulla tavola periodica.
Esempio. Calcolare la densità teorica del rame (Cu) con struttura c.f.c. , Peso
Atomico 63,5 g/mol. e raggio atomico 0,128 nm.
n= 4 ; ACu= 63,5 g/mol ; NA= 6.023 x 1023 atomi/mol ; V c.f.c. = 16 R321/2
r = (4x63,5)/ 16 (1,28x10- ) 2
8 3 1/2x6.023
x 1023 = 8,89 g/cm3
STRUTTURE COMPATTE CFC E EC
Impilamento di piani di massimo
Impacchettamento atomico
CFC
SEQUENZA DI IMPILAMENTO
ABCABCABCABC…..
Nella cella elementare CFC i piani a
massimo impacchettamento atomico
corrispondono ai piani atomici 111
Metalli CFC : Al, Cu
HCP
Sequenza di impilamento di piani a massimo
impacchettamento atomico
ABABABABAB…….
Nella cella esagonale i piani a massimo
impacchettamento atomico sono i piani basali
ed hanno indici di Miller 0001
Appartengono a questa classe Cd, Mg, Zn, Ti …
ALLOTROPIA
Un materiale solido può assumere più di una forma cristallina e tale fenomeno è
detto allotropia. Un esempio di allotropia ci è dato dal carbonio. Il carbonio può
formare 4 diverse strutture solide ordinate.
Il diamante e la grafite sono forme allotropiche del carbonio, queste due strutture
sono caratterizzate da differenti tipi
di legame fra gli atomi di carbonio. Inoltre esistono anche i fullereni e i nanotubi in
cui un gran numero di atomi da origine a forme sferiche e cilindriche,
rispettivamente.
ISOTROPIA/ANISOTROPIA
Direzioni diverse in un cristallo hanno diverso impacchettamento. Per esempio gli
atomi lungo gli spigoli della cella elementare FCC sono più lontani di quelli lungo la
diagonale delle facce. Questo causa anisotropia nelle proprietà del cristallo. Ad
esempio la deformazione dipende dalla direzione in cui lo sforzo è applicato.
METALLO
MODULO DI
[100]
ELASTICITA’
[110]
ALLUMINIO
63,7
72,6
76,1
RAME
66,7
130,3
191,1
FERRO
125,0
210,5
272,7
[111]
In alcuni materiali policristallini l’orientazione dei diversi grani è random , pertanto
anche se la cella elementare presenta anisotropia il solido nel suo complesso risulta
isotropo. Nel caso in cui i grani del materiale policristallino abbiano un orientazione
preferenziale le proprietà del solido possono conservare una certa anisotropia.
Scaricare