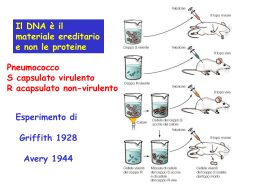
UN PO’ DI STORIA... 1900 - Riscoperta delle leggi della trasmissione ereditaria dei caratteri, che Mendel aveva pubblicato nel 1865 e che avevano come assunto l'esistenza di fattori discreti, elementen, i geni, che determinano i caratteri e che si trasmettono da una generazione all'altra. 1902 - Sutton ipotizza che i fattori mendeliani, i geni, siano localizzati sui cromosomi. 1910-1911 - Morgan inizia a mappare i geni sui cromosomi del moscerino della frutta D.Melanogaster 1944 - Avery MacLeod e McCarty stabiliscono che il DNA è il materiale ereditario. 1953 - Watson e Crick propongono la struttura a doppia elica per il DNA 1956 - Viene stabilito che il patrimonio cromosomico completo dell'uomo è di 46 cromosomi. 1961-1966 - Viene decifrato il codice genetico, ovvero stabilito il rapporto tra le 64 triplette possibili a partire dalle 4 basi nucleotidiche del DNA, e i 20 aminoacidi che formano le proteine. 1967 - Mary Weiss e Green introducono la tecnica dell'ibridazione delle cellule somatiche che rende più agevole la mappatura dei geni umani. 1972 - Berg costruisce la prima molecola DNA ricombinante in vitro utilizzando gli enzimi di restrizione. Vengono realizzati con successo i primi esperimenti di clonazione del DNA. 1973 Cohen, Chang e Boyer costruiscono il primo batterio ricombinante. Si tiene la prima conferenza sulla - mappatura dei geni umani. 1974 - Cohen e Boyer ottengono l'espressione di un gene estraneo trapiantato in un batterio con la tecnica del DNA ricombinante. 1975 - Si tiene la conferenza di Asilomar che propone una moratoria sugli esperimenti con il DNA ricombinante in assenza di linee guida per la sicurezza delle manipolazioni genetiche. 1977 - Per la prima volta un gene umano viene ricombinato e inserito in un batterio per clonare una proteina, la somatostatina Sanger, già premio Nobel come inventore del metodo per sequenziare le proteine, sviluppa un metodo nuovo ed efficiente per sequenziare il DNA (contemporaneamente Gilbert e Maxam inventano un metodo analogo). 1978 - Boyer costruisce una versione sintetica del gene dell'insulina e lo inserisce in un batterio. La Genentech, fondata nel 1976 dallo stesso Boyer insieme a Swanson, otterrà il permesso di commercializzare l'insulina prodotta per ingegneria genetica nel 1982 e quindi il brevetto 1978-1980 - Vengono sviluppate nuove tecniche basate sull'ibridazione molecolare e l'utilizzazione come marcatori delle variazioni individuali del DNA per mappare fisicamente i geni e le sequenze geniche sui cromosomi. Grazie alle nuove tecniche e alla collaborazione internazionale tra i mappatori, i geni mappati passano da 579 nel 1981 a 1.879 nel 1991: tra questi vengono mappati il gene della corea di Huntington (1983), il gene per la distrofia muscolare (1987) e il gene per la fibrosi cistica (1989). 1980 - Viene concesso il primo brevetto su una forma di vita geneticamente modificata, un microrganismo che si nutre di petrolio. Mullis inventa la tecnica della PCR. 1981 - Viene prodotto il primo animale transgenico. 1983 - Viene inventato il primo sequenziatore automatico. 1989 - Viene creato il National Center for Human Genome Research (NCHGR), guidato da James Watson per mappare e sequenziare tutto il DNA umano entro il 2005 1992 - Craig Venter crea l'Institute for Genomics Research. 1993 - Francis Collins assume la direzione del National Human Genome Research Institute (ex NCHGR). 1995 - Viene pubblicata la prima sequenza completa del genoma di un organismo vivente diverso da un virus, il batterio Haemophilus influenzae. 1996 - Viene riportato il sequenziamento completo del primo organismo complesso, il lievito Saccharomyces cerevisiae. 1997 - Viene costruito il primo cromosoma umano artificiale e annunciata la clonazione di un mammifero, Dolly . 1986 - Viene proposto da diversi biologi molecolari, tra cui Renato Dulbecco, di sequenziare completamente il genoma umano. 1988 - Viene concesso il brevetto per un topo transgenico altamente suscettibile al tumore del seno. 1998 - Viene pubblicata una prima bozza della mappa del genoma umano, che mostra la localizzazione di più di 30.000 geni. Viene pubblicata la sequenza completa del primo genoma di un animale, il verme Caenorhabditis elegans. Venter e la Perkin Elmer Corporation fondano la Celera Genomics con l'intento di sequenziare tutto il genoma umano in tre anni con il metodo del whole genome shotgun. 2000 - Viene pubblicata la sequenza completa del genoma di Drosophila melanogaster e viene annunciato dalla Celera Genomics il sequenziamento di tutte le basi nucleotidiche del DNA umano con il metodo whole genome shotgun. Date storiche della sequenza del DNA Avery propone il DNA come materiale genetico 1944 1869 1953 Holley sequenza il tRNA di lievito 1965 1970 METODO SANGER (terminazione di catena) METODO MAXAM-GILBERT (degradazione chimica) KARY MULLIS Introduce la PCR 1977 1980 1986 Automazione completa Sequenziamento completo del genoma umano 1989 2000 Miescher osserva per la prima volta il DNA Watson e Crick determinano la struttura della doppia elica Vengono sviluppate le tecniche per la sintesi degli oligonucleotidi e per la degradazione chimica del DNA. Viene introdotta l’elettroforesi su gel per la Separazione di frammenti di DNA Il DNA genomico viene clonato nel fago M13 o in vettori plasmidici, nascono i primi programmi informatici di analisi dei dati, vengono sviluppate nuove tecnologie per il sequenziamento AUTOMAZIONE PARZIALE Vengono sviluppate le prime apparecchiature per il sequenziamento che impiegano sistemi per la rilevazione della fluorescenza. Sequenziamento Estrazione di DNA o RNA PCR o amplificazione mediante clonaggio Purificazione del campione di DNA Cycle Sequencing Precipitazione dei prodotti di cycle sequencing Sequenziamento automatico Analisi dei dati Sequenziamento del DNA Metodi tradizionali ed odierni Metodi di Maxam-Gilbert e di Sanger Hanno in comune la generazione di una scaletta di frammenti di DNA A singolo filamento, ciascuno più lungo del precedente di una base. Frammenti di dimensioni crescenti possono essere separati mediante Corsa elettroforetica su gel di poliacrilammide. A corsa ultimata il gel viene posto a contatto con una pellicola radiorafica Sulla quale lascia impressa la disposizione delle bande. L’immagine ottenuta, letta di seguito darà la sequenza delle basi. Il metodo Maxam-Gilbert (o metodo di sequenziamento a degradazione chimica) Questo metodo, basato sulla degradazione chimica di frammenti di DNA a doppio filamento, è ormai poco utilizzato, perché non adatto al sequenziamento automatico e perché utilizza composti chimici dannosi alla salute Si differenzia da tutti gli altri metodi perché non utilizza sintesi enzimatiche ma trattamenti chimici che agiscono a livello di singoli nucleotidi. I frammenti di DNA a doppio filamento possono essere generati per frammentazione o digestione enzimatica. In entrambi i casi le molecole a doppio filamento devono essere convertite in molecole a singolo filamento e marcate terminalmente Marcatura terminale al 5’ o al 3’ del DNA a doppio filamento Denaturazione e separazione dei due filamenti Il DNA a singola elica viene suddiviso in quattro campioni, ognuno dei quali viene trattato con un reagente chimico che demolisce una o due delle 4 basi del DNA. G G+A C+T C = DMS + piperidina = DMS + piperidina + acido formico = idrazina + piperidina = idrazina + piperidina in NaCl 1,5 M Le reazioni sono controllate in modo da avere una frammentazione parziale: statisticamente tutte le possibili basi saranno degradate producendo una serie di frammenti la cui lunghezza dipenderà dalla distanza tra l’estremità marcata e il sito di taglio Separazione dei frammenti marcati mediante gel elettroforesi e Visualizzazione dei risultati mediante autoradiografia Il metodo Maxam-Gilbert Il metodo Sanger (o metodo a terminazione di catena) Il sequenziamento a terminazione di catena coinvolge la sintesi di nuovi filamenti di DNA, complementari ad uno stampo a singolo filamento Per la sintesi del DNA si utilizzano DNA polimerasi caratterizzate da •Elevata processività •Bassa attività esonucleasica 5’-3’ •Bassa attività esonucleasica 3’-5’ (es. Klenow, Sequenasi) Il metodo Sanger (o metodo a terminazione di catena) Nel sequenziamento a terminazione di catena la reazione oltre ad Uno stampo a singola elica ha bisogno di: un innesco specifico (primer) La marcatura con un dNTP* marcato, di solito con 35S Sintesi del filamento marcato PRIMER DNA Polimerasi 5’-A T C T T T T A G A G T A C C 3’-T A G A A A A T C T C A T G G A C T C T C T A C T A T C T A C A T G T A -5’ PRIMER DNA Polimerasi 5’-A T C T T T T A G A G T A C C T G A G*A G A T G A T A G*A 3’-T A G A A A A T C T C A T G G A C T C T C T A C T A T C T A C A T G T A -5’ Il metodo Sanger (o metodo a terminazione di catena) Nel sequenziamento a terminazione di catena la reazione oltre ad • uno stampo a singola elica • un innesco specifico (primer) • la marcatura con un dNTP* marcato, di solito con 35S ha bisogno di: • una miscela di ddNTP, uno per ogni reazione di sequenza Terminazione della catena La sintesi del filamento complementare, tuttavia non prosegue indefinitamente, perché la miscela di reazione contiene, in quattro distinte reazioni piccole quantità di specifici dideossi nucleotidi trifosfati, ddATP, ddTTP, ddCTP e ddGTP, che bloccano l’allungamento perché posseggono un solo atomo di idrogeno al posto del gruppo -OH in 3’ PRIMER 5’-A 3’-T T C T T T T A G A G T A C C T G A G*A G A T G A T A G*A DNA Polimerasi A G A A A A T C T C A T G G A C T C T C T A C T A T C T A C A T G T A -5’ + ddNTP ( per es. ddCTP) STOP 5’-A 3’-T T C T T T T A G A G T A C C T G A G*A G A T G A T A G*A T G T AddC A G A A A A T C T C A T G G A C T C T C T A C T A T C T A C A T G T A -5’ Il risultato è una serie di frammenti interrotti ciascuno in corrispondenza di ogni dCTP ddCTP ddCTP ddCTP ddCTP ddCTP ddCTP Schema di sequenziamento a terminazione di catena DNA stampo a singola elica 3’-GGCTAAC 5’ Ibridazione con Il primer 3’ 3’-GGCTAAC + [35S]dATP+dCTP,dGTP,dTTP(dNTP)+Sequenasi ddATP, dNTP ddCTP, dNTP -CCG ddA -ddC -C ddC A C ddGTP, dNTP ddTTP, dNTP -CC ddG -CCGATT ddG G -CCGA ddT -CCGAT ddT T -CCGATT ddG -CCGAT ddT -CCGA ddT -CCG ddA -CC ddG -C ddC -ddC G T T A G C C Sequenza: 5’-CCGATTG Direzione di lettura Sequenziamento manuale Pratica ormai superata dal seq. automatico Marcatura Radioattiva Primer marcati con 32P oppure 35S dNTP Colorazione Silver Staining Evidenzia direttamente su gel le bande senza ulteriori modificazioni, attraverso la precipitazione di Ag Nuovi metodi di sequenziamento Il sequenziamento a ciclo termico ( PCR asimmetrica) Il sequenziamento automatizzato con marcatori fluorescenti Il pirosequenziamento Sequenziamento automatico 1 Colorante - 4 Corsie Per ogni campione si eseguono 4 reazioni di estensione separate (una per ogni ddNTP) utilizzando un unico colorante L’uso di 4 corsie aumenta però la variabilità di corsa Sequenziamento automatico 4 Coloranti - 1 Corsia Unica reazione ed unica corsa; Ad ogni nucleotide è associata una diversa colorazione: ddATP ddTTP ddGTP ddCTP Questo sistema aumenta la produttività ed elimina la variabilità di corsa Il sequenziamento automatizzato con marcatori fluorescenti Coniugando a ciascun ddNTP un diverso marcatore fluorescente, è possibile effettuare le quattro reazioni di sequenziamento in un unico tubo da saggio e caricare il tutto in un solo pozzetto di gel ddA ddT ddC ddG Sequenziamento automatico Metodi di Marcatura Marcatura dei Primers primers progettati con “tag” colorati a: Fluorescenza UV Fluorescenza IR minore Background maggiore Sensibilità Marcatura dei Terminatori ddNTPs marcati con fluorofori o differenti coloranti Sequenziamento automatico Terminatori BigDye Sistemi a trasferimento di energia a singola molecola, costituiti da accettore e donatore legati da un linker Vantaggi: Segnale omogeneo, basso rumore di fondo, luminosità maggiore, facilità interpretativa per A e G attigue D A Dalla cycle sequencing all’elettroforetogramma... Le emissioni fluorescenti vengono captate da un rilevatore e le informazioni vengono integrate e trasformate in picchi di colore diverso, con aree proporzionali all’intensità di emissione. Sequenziamento automatico Prevede l’utilizzo di marcatura a fluorescenza rilevata automaticamente da un laser Sequenziatori a Gel reazione e caricamento dei campioni sono eseguite manualmente Esistono kits in commercio che permettono di velocizzare e standardizzare tali operazioni fornendo mix di reazione, soluzioni di poliacrilammide a cui aggiungere soltanto Temed o gel preformati Gel per elettroforesi “slab” gel di poliacrilammide (dal 4% al 6%) molto sottili in condizioni denaturanti (Urea) Apparati verticali di lunghezza variabile a seconda del numero di basi da leggere (22 cm-1 m) Il forte campo elettrico a cui vengono sottoposti produce elevato calore che deve essere dissipato con piastre di alluminio o ventole Sequenziamento automatico Sequenziatori Capillari l’intervento dell’operatore è minimo il sistema carica automaticamente il capillare con il polimero di corsa esegue la separazione elettroforetica i frammenti marcati di DNA vengono rilevati man mano che corrono lungo il capillare Sequenziamento automatico Sequenziatori Capillari 1 Capillare 16 Capillari 96 Capillari (Scala di Produzione) Richiede campioni privi di: sali, molecole cariche negativamente, proteine, detergenti e templates non marcati Sequenziamento automatico Sistemi di Rilevamento Camera CCD ( Charge - Coupled Device) Altissima risoluzioneImmagine Elettroferogramma Sequenza in pochi minuti Camera a Lettura Multipla Assenza di Filtri Uso di qualsiasi colorante Doppio Laser Doppio Rilevatore Es. NIR o SBS Sequenziamento automatico Software Controlla e ottimizza i parametri elettroforetici Normalizza le bande Sottrae il background Corregge l’effetto “Smile” Adatta gli algoritmi se mobilità e spaziatura dei picchi escono dal range Reazione fallita: non presenta picchi definiti e ha un alto rumore di fondo Possibili Cause Possibili Soluzioni • Il primer non trova sito di annealing • Cambiare primer • Il DNA presenta contaminazioni di vario tipo • Ripreparare il DNA • La quantità di DNA e' insufficiente • La quantià di primer è insufficiente • Aumentare la quantità di DNA • Aumentare la quantità di primer • Il primer è stato disegnato male, es. temperatura di melting troppo bassa • Ridisegnare il primer Campione che presenta rumore di fondo sufficientemente alto da causare ambiguità (N) nel riconoscimento dei picchi da parte del software Possibili Cause Possibili Soluzioni • Aumentare la quantità di DNA • La quantità di DNA e' insufficiente e il segnale troppo debole • Purificare meglio il DNA • Il DNA presenta contaminazioni che inibiscono la reazione • Ci sono altri templati contaminanti • Ripreparare il DNA Perdita di risoluzione precoce per cui i picchi sono sempre meno definiti Possibili Cause • Il DNA presenta contaminazioni che inibiscono la reazione Possibili Soluzioni • Purificare meglio il DNA Sequenza doppia dopo un tratto buono Possibili Cause • Clone o PCR multiple con stesso tratto iniziale, es. vettore Possibili Soluzioni • Ripreparare DNA in modo da avere un tipo di templato unico • Siti multipli di attacco del primer sul DNA • Mutazione frame shift • Slittamento dopo una regione omopolimerica • Cambiare primer Sequenza fuori scala Possibili Cause • Troppo DNA • Struttura secondaria che blocca la reazione Possibili Soluzioni • Riquantizzare e ridurre la quantità di DNA • Pretrattare con DMSO Regione ricca in GC Possibili Cause • Struttura secondaria che impedisce l'avanzamento della polimerasi perchè, per effetto dell'alta Tm del tratto di DNA, i due filamenti non si separano bene Possibili Soluzioni • Sequenziare un prodotto di PCR amplificato sostituendo il 75% del dGTP con 7-deaza-dGTP • Modificare il ciclo standard di sequenziamento con temperature più alte Sequenziamento automatico: Applicazioni in ambito biomedico Sequenziamento di DNA o cDNA sconosciuto ( es. per caratterizzare regioni del DNA e trascritti genici precedentemente localizzate con analisi di linkage) • Primer walking • Shotgun sequencing utilizzo di vettori di clonaggio • Vettori fagici a singolo filamento • Vettori plasmidici a doppio filamento • Vettori fagomidici • Vettori per grossi inserti di DNA Sequenziamento di DNA o cDNA conosciuto per analisi mutazionale utilizzo di PCR Mapping Tecnica del “Chromosome Walking” Sequenziamento di Library Progetto Genoma Sequenziamento Ex Novo Uso di Primers Universali Es. M13 Tecnica del “Random Sequencing” Sfrutta l’Overlapping dei vari frammenti Assemblaggio ed Allineamento delle sequenze tramite Computer Le finalità principali del progetto genoma riguardano l'acquisizione i informazioni utili per individuare l'eventuale implicazione di alterazioni nella sequenza del DNA nello sviluppo di patologie genetiche nell'uomo, e per comprendere le basi genetiche dell'evoluzione e del funzionamento dell'organismo umano Sintesi di Oligonucleotidi analisi dell’espressione genica Probe per Microarray farmacogenomica SNPs Oligonucleotidi antigene/antisenso Dare un senso ai dati di sequenza non è facile!! Isole CpG: la citosina può essere metilata nei geni inattivi Sequenze di siti di splicing in 5’ e 3’: come marcatori per gli introni e le zone di confine con gli esoni ORF: mRNA CUUAGCGUAGCUACUAGACUAG fase 1 CUU/AGC/GUA/GCU/ACU/AGA/CUA/G fase 2 CU/UAG/CGU/AGC/UAC/UAG/ACU/AG fase 3 C/UUA/GCG/UAG/CUA/CUA/GAC/UAG Open reading frame Perché sequenziare anche organismi diversi da Homo Sapiens? Ad esempio per confrontare le sequenze altamente conservate, è più probabile che codifichino proteine funzionalmente importanti Sequenziamento di DNA o cDNA sconosciuto ( es. per caratterizzare regioni del DNA e trascritti genici precedentemente localizzate con analisi di linkage) • Primer walking • Shotgun sequencing utilizzo di vettori di clonaggio • Vettori fagici a singolo filamento • Vettori plasmidici a doppio filamento • Vettori fagomidici • Vettori per grossi inserti di DNA Sequenziamento di DNA o cDNA conosciuto per analisi mutazionale utilizzo di PCR Comparativa e/o Mutazionale Uso Diagnostico Applicazione non di routine Monitoraggio pazienti in terapia Test di Genotyping Es. HIV Implicazioni Terapeutiche Dimensione della mutazione Poliploidie Genoma Cromosoma Gene Aneuploidie Bandeggio cromosomico Fish di interfase Riarrangiamenti cromosomici Fish cromosomica Piccole delezioni/duplicazioni Delezioni/duplicazioni geniche Genetica molecolare Delezioni/duplicazioni nucleotidiche Mutazioni nucleotidiche Sequenziamento Mutazione di un singolo nucleotide Mutazioni per inserzione o delezione di nucleotidi in eterozigosi Sequenziamento del tratto nucleotidico delle immunoglobuline (Ig) relativo al riarrangiamento della regione variabile CDR3 specifica del clone tumorale, da utilizzare in neoplasie linfoidi di tipo B per il monitoraggio della malattia minima residua e come base per la produzione di vaccini anti-idiotipici paziente-specifici. Sequenziamento della VH-CDR3 Pession et al, Leuk Lymphoma 2003 Sequenziamento regione CDR3 del TCR per caratterizzazione cloni di linfociti T in vitro/vivo CDR 3 REGIONI CHE DETERMINANO LA COMPLEMENTA RIETA’ Sequenziamento regione CDR3 del TCR per caratterizzazione cloni di linfociti T in vitro/vivo Montagna et al int.j of cancer 2004 Genotipizzazione del genoma virale del lentivirus HIV, responsabile della sindrome da immunodeficienza acquisita umana (AIDS) una delle caratteristiche di questo virus è quella di andare in contro spesso a mutazioni casuali nel proprio genoma. ne consegue la selezione di ceppi resistenti ai farmaci HIV ViroSeq Kit (Applied Biosystems Inc.) Individuazione Resistenze farmaci in pazienti non rispondenti TRUGENETM HIV-1 Genotyping Kit (Visible Genetics Inc) HIV Software allinea sequenza con WT Rileva Mutazioni Regole che correlano singole o combinazioni di mutazioni al grado di resistenza per ogni farmaco Accesso a Banca Dati Valutazione dello stato di metilazione di un gene ed in particolare del suo promotore Tecnica del bisufilto Il trattamento con bisulfito converte le citosine non metilate in timine Le citosine metilate (quelle delle isole CpG) rimangono tali Attenzione nel progettare i primers !! (escludere le parti con CpG) Decitabina: demetilante inibendo la DNA metil-transferasi LAM in particolare con riarrangemento di MLL Dott.ssa Formica, dott.ssa Dan, lab. Oncoped. Elenco dei siti che contengono informazioni sul Progetto Genoma Umano e sui frammenti di DNA sequenziati. The Huma n Genome Project http://www.nhgri.nih.gov un sito dedicato al Progetto Genoma Umano gestito dal NHGRI (The National Huma n Genome Research Institute) GenBank http://www.ncbi.nlm.nih.gov un sito in cui si possono trovare sequenze di DNA e di proteine. EMBL http://www.ebi.ac.uk GDB (genome database): http://www.gdb.org/ dbEST http://www.ncbi.nlm.nih.gov/dbEST/i ndex.html OMIM (on-l ine Mendelian Inheritance in Man) accesso attraverso GDB MGD (Mouse Genome D ata base) http://www.informa tics.jax.org un sito dove sono raccolte le sequenze del genoma umano. il più impo rta nte database concernente la mappatura del genoma . raccoglie le sequenze parziali ottenute dai cDNA catalogo elettronico di Mendelian Inheritance in Man di Victo McK usick, un elenco delle ma lattie umane ereditarie. database del genoma mu rino, Généthon CHLC (Co-operative Huma n Linkage Center) CEPH CELERA bibliografia sul progetto genoma umano Whitehead Institute http://www.genethon.fr/ http://lpg.nci.nih.gov/CHCL/ http://www.cephb.fr/bio/cephgenethon-map.html http://www.celera.com/ mappa genetica basata sui marcatori con ripetizioni (CA)n database contenente una mappa genetica e dati riguardanti il genotipo e i marcatori genetici, contenente un programma p er l’analisi di linkage mappa fisica del genoma umano basato sugli YAC. il sito internet della società privata americana diretta da Greg Venter www .nature.com/genomics/0 http://www-genome.w i.mi t.edu/ mappa fisica del genoma umano basata sugli YAC Il clonaggio dell’intero genoma umano non è difficile tecnicamente e sono disponibili genoteche in fagi od in cromosomi artificiali di lievito abbastanza ampie da contenere diverse volte il genoma umano; ma il DNA di queste genoteche è stato clonato in modo casuale dal genoma. Perché abbia un senso il DNA in questi cloni deve essere strutturato nell’ordine corretto, in modo da poter ricostruire il genoma , o suoi frammenti che siano abbastanza ampi da contenere porzioni di sequenze biologicamente interessanti. Sospetto clinico (sintomatologia, storia familiare) Isolamento dal sangue periferico di linfociti circolanti Analisi del cariotipo Estrazione del DNA Amplificazione tramite PCR Southern blot Corsa su gel Diagnosi citogenetica SSCP RFLP sequenziamento Creazione o distruzione di un sito di restrizione diagnosi di delezioni inserzioni duplicazioni identificazione di nucleotidi alterati diagnosi di specifiche mutazioni puntiformi
Scaricare