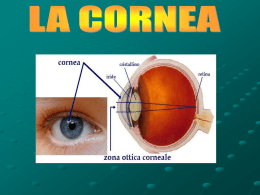
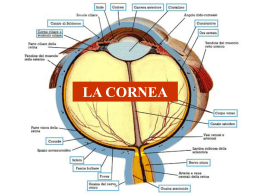
PATOLOGIE CORNEALI Corso: Malattie Apparato Visivo UNIVERSITA’di BOLOGNA Facoltà di Medicina e Chirurgia CORSO DI LAUREA IN INFERMIERISTICA. PROF. SERGIO ZACCARIA SCALINCI Malattie congenite della cornea Le malattie corneali congenite si manifestano: • isolatamente • associate (vista l’origine embrionaria comune di parecchie strutture) Organogenesi della cornea Durante tutte le fasi di differenziazione, il tessuto corneale è sempre avascolare. La reale trasparenza corneale inizia verso il quarto mese con un progressivo arricchimento in cheratosolfato che compare nel mesenchima corneale, espressione di un differenziamento metabolico dei cheratociti embrionali. CLASSIFICAZIONE EMBRIOLOGICA 1. Anomala migrazione delle cellule della cresta neurale: Sclerocornea, Embriotoxon posteriore, Sindrome di Axenfeld e di Rieger, Anomalia di Peters, Glaucoma Congenito 2. Abnorme proliferazione delle cellule della cresta neurale Atrofia essenziale dell’iride, sindrome di Chandler, Sindrome di Cogan 3. Anomala differenziazione delle cellule della cresta neurale: Distrofia corneale posteriore polimorfa, cornea guttata congenita, distrofia di Fuchs. CLASSIFICAZIONE MORFOLOGICA A) Anomalie di grandezza e forma (Criptoftalmo, assenza della cornea, megalocornea, microcornea, cornea piana, cornea ovale, megaloftalmo anteriore, cheratoglobo) B) Rimaneggiamenti strutturali (Sclerocornea, Sindrome di Peters, stafiloma corneale anteriore, embriotoxon anteriore o arco iuvenilis) C) Displasie marginali posteriori (cheratocono posteriore, embriotoxon posteriore di Axenfeld, sindrome di Rieger, disginesia mesodermica, cheratoectasia) D) Distrofie corneali eredo-familiari A) O CRIPTOFTALM Eccezionale Mono o bilaterale La pelle del viso sostituisce le palpebre e ricopre il cavo orbitario nascondendo l’occhio Può essere associato a discefalia, sindattilia, anomalie urogenitali Trasmissione a volte ereditaria, autosomica recessiva. A) ASSENZA DELLA CORNEA Rara Casi con assenza completa dell’invaginazione ectodermica (cornea, C.A., cristallino); occhio costituito solo da vescicola rivestita da tessuto simil sclerale con, all’interno, tessuto retinico anomalo. A) MICROCORNEA •Mono o bilaterale, senza alterazioni della trasparenza. (Se le dimensioni dell’occhio e quelle della cornea sono diminuite contemporaneamente, ma i tessuti sono normali, si parla di nanoftalmia. Se invece si associano alterazioni si parla di microftalmo). •Ereditaria, autosomica dominante •Diametro orizzontale sotto i 9mm alla nascita •Arresto di sviluppo al IV-V mese di vita fetale •Il difetto refrattivo associato è l’ipermetropia elevata •Può associarsi ad anomalie del segmento anteriore (ipertonia, cataratta, coloboma). •Col tempo può indursi o un glaucoma ad angolo aperto o un glaucoma acuto da blocco dell’angolo. A) MICROCORNEA •La microcornea è presente in molte anomalie sistemiche: s. di WeilMarchesani, di Rieger e di Ehlers-Danlos, le sindromi cromosomiche di Turner, di De Grouchy, malattia Di Norrie, di Meckel, di SjogrenLarrson, la trisomia 13 e 15, alcune malformazioni cranio-facciali ed alcune malattie neurologiche. •diagnosi differenziale con microftalmia o nanoftalmia tramite misurazione delle dimensioni del bulbo con l’ecografia. A) CORNEA OVALE Avanzamento della sclera sul margine superiore ed inferiore o sui margini laterali della cornea La cornea ovale orizzontale è frequente nella sclero cornea La cornea ovale verticale spesso è presente nella microcornea con sdr. Di Turner o esito di cheratite prenatale luetica. A) CORNEA PLANA Disgenesia con raggio di curvatura tra 20 e 30 D Può essere totalmente piatta con scomparsa dell’angolo sclerale Limbus neovascolarizzato Refrazione ipermetropica Visus ridotto (coesistenza di altre anomalie del segmento anteriore) Eredità autosomica dominante recessiva Associata all’osteogenesi imperfetta o alle A) MEGALOCORNEA •Bilaterale, simmetrica, alla nascita diametro corneale già da adulto: 11-12mm •Spessore normale, soprattutto uomini per l’ereditarietà legata al sesso •E’ a volte associata ad altre alterazioni del segmento anteriore. La cornea risulta chiara,a spessore normale , senza pieghe della membrana di Descemet e PIO nella norma. Può presentare dei depositi. •Associato ± ad arco giovanile, fuso di Krukenberg, lussazione della lente, sdr. Di Marfan, craniosinostosi, ect. A) MEGALOCORNEA •diagnosi differenziale con glaucoma congenito e cheratoglobo. •E’ una malformazione nel complesso benigna, compatibile con una buona funzione visiva, soprattutto in rapporto al difetto refrattivo concomitante (miopia, astigmatismo). A) MEGALOFTALMO ANTERIORE Aumentata dimensione della cornea e di tutto il segmento anteriore Associata sovente ad iridodonesi, facodonesi con lussazione del cristallino e glaucoma secondario Presente nella sdr. di Marfan, mucopolisaccaridosi II, sdr. di Apert. A) O CHERATOGLOB Bilaterale Cornea grande, trasparente e globosa che protrude in avanti Assottigliata, soprattutto in periferia Sopra i 16mm di diametro, cheratometria superiore a 50D Trasmissione autosomica recessiva (arresto di sviluppo posteriore) Asintomatica Frequente associazione con la sclera blu e l’iperestensibilità delle articolazioni mano-caviglia A) BUFTALMO Il glaucoma congenito non è una patologia corneale, ma le sue conseguenze su di essa sono di notevole importanza. Esordio: alla nascita 40% entro primo anno di vita 86% anche diversi anni dopo (glaucoma infantile) 14% Il primo segno di un glaucoma congenito è la presenza di una cornea di grandi dimensioni nel corso dei primi 6 mesi di vita. Nei casi più gravi la cornea è opalescente. A) BUFTALMO Durante la visita, in anestesia generale, si nota allargamento del limbus, cornea con diametro superiore a 11 mm, edema epiteliale, rottura della membrana di Descemet parallele al limbus. La camera anteriore è profonda e l’iride poco pigmentata, La IOP è superiore a 14 mmHg sotto anestesia. Rottura Descemet B) SCLEROCORNEA •Deriva da un arresto nella differenziazione limbare durante la migrazione delle cellule della cresta neurale. •bilaterale nel 90% dei casi, non progressiva •ereditarietà sporadica oppure autosomica dominante o recessiva. •2 tipi: periferica e totale (opacità diffusa con un disco centrale opalescente circondato da una zona totalmente opaca e vascolarizzata. Il limbus è male riconoscibile, il parenchima è sostituito da tessuto sclerale opaco in cui si notano fini arcate vascolari). B) SCLEROCORNEA • Spesso associata a brachicefalo, ipoacusia, trisomia 18, sclera blu. • Diagnosi differenziale con il leucoma corneale. • Il visus è di solito molto basso, associato a nistagmo. Aspetto istologico piani profondi Aspetto istologico piani superficiali Aspetto ultrastrutturale stroma anteriore B) ANOMALIA DI PETERS Deriva da una anomala migrazione della cellule della cresta neurale o da una sublussazione della lente durante la vita intrauterina con conseguente alterazione anche del foglietto endoteliale. ANOMALIA DI PETERS Questa anomalia è bilaterale nell’80% dei casi. Comporta classicamente un leucoma corneale centrale o paracentrale, sinechie iridee anteriori e opacità sottocorticale anteriore del cristallino. ANOMALIA DI PETERS Può manifestarsi anche come leucoma avascolare che protrude dal limbus verso il centro o come una massa rilevata simile ad un dermoide o un arco biancastro paracentrale parallelo al limbus. In alcuni casi il cristallino è aderente alla cornea o sublussato posteriormente. Si associa frequentemente a glaucoma secondario per le sinechie anteriori periferiche, microftalmia, microcornea, cornea plana, coloboma, cataratta. L’associazione con patologie sistemiche non è frequente. Diagnosi differenziale con microftalmia, glaucoma congenito e leucoma B) STAFILOMA ANTERIORE malformazione mono o bilaterale, totale, del segmento anteriore con cornea che protrude tra le palpebre per glaucoma secondario La cornea è opaca e vascolarizzata, l’iride aderente alla cornea, il cristallino opaco, rimpicciolito o assente. L’occhio è microftalmico senza possibilità funzionale Possibili infezioni gestazionali B) EMBRIOTOXON ANTERIORE O ARCO JUVENILIS Opacità bianca anulare parallela al limbus nel parenchima superficiale Fosfolipidi, colesterolo, trigliceridi Può associarsi a megalocornea, sclera blu, aniridia C) CHERATOCONO POSTERIORE CIRCOSCRITTO Anomalia congenita rara, caratterizzata da un’indentazione conica centrale della superficie corneale posteriore con diminuzione dello spessore dello stroma. La superficie corneale anteriore è di solito normale. Patologia unilaterale e sporadica nella maggior parte dei casi Normalmente l’acuità visiva non è compromessa C) CHERATOCONO POSTERIORE CIRCOSCRITTO assottigliamento corneale, assenza della m. basale dell’epitelio, sostituzione della Bowman con tessuto fibroso, Descemet lesionata ed irregolare. Etiopatogenesi: incerta (probabile ritardo di separazione della vescicola lenticolare dall’ectoderma oppure iposviluppo dell’endotelio nella zona centrale con ostacolo alla migrazione e distribuzione delle cellule mesodermica nello stroma oppure alterazioni primitive della Descemet). Può essere associata a: lenticono anteriore, aniridia, anello di Fleisher, ectropion uveae; ipertelorismo, brachidattilia, ritardo mentale. C)EMBRIOTOXON POSTERIORE O DISPLASIA MERGINALE POST DELLA CORNEA Anomalia ereditaria occasionale Costituita da stria filiforme anulare, parallela al limbus, aderente alla linea di Schwalbe, ed un pò protrudente in C.A. Può essere associata a cornea plana, ectopia o aniridia c) Sindrome di Axenfeld Deriva da una anomalia di migrazione delle cellule della cresta neurale 1. Anomalia di Axenfeld: Embriotoxon posteriore associato a processi iridei prominenti e goniosinechie 2. Sindrome di Axenfeld: anomalia di Axenfeld associata a glaucoma 3. Ereditarietà autosomica dominante 4. Associata ad alterazioni scheletriche, quali ipertelorismo ed asimmetria faciali o ad altre sindromi sistemiche (Sindrome di Marfan, Lowe, Turner) C) Sindrome di Rieger 1. Anomalia di Rieger: • Anomalia nella prima e nella terza fase di migrazione mesenchimale durante lo sviluppo embrionale • Embriotoxon posteriore associato a processi iridei prominenti e con sinechie anteriori periferiche ed ipoplasia o atrofia dello stroma irideo. • Frequenti piccole opacità corneali periferiche a livello della Descemet, più raramente cheratocono posteriore. • Pupilla con anomalie di forma, dimensione e posizione, talora multipla • Molto frequente è la comparsa di glaucoma C) Sindrome di Rieger 1. Sindrome di Rieger: • E’ caratterizzata dall’associazione dell’anomalia di Rieger con malformazione cranio-facciali come la radice del naso larga e piatta, l’ipoplasia mascellare, la microdonzia o l’anodonzia e alterazione degli arti e della colonna vertebrale, la sordità, il ritardo mentale e la sindrome di Marfan. D) DISTROFIE CORNEALI 1.Distrofie anteriori Le distrofie corneali sono un gruppo di malattie progressive, bilaterali, nella maggioranza dei casi geneticamente determinate, che inducono opacità corneale in assenza di infiammazione e, molto raramente, presenti alla nascita soprattutto nelle forme posteriori ma per lo più ad espressione fenotipica tra la Iª e la IVª decade di vita. • microcistica di Cogan • Reis-Bücklers • Meesmann • Schnyder 2. Distrofie stromali • a lattice I, II, III • granulare I, II, III • maculare I, II 3. Distrofie posteriori • endoteliale (Fuchs) • polimorfa posteriore DEG. MICROCISTICA DI COGAN O DISTROFIA DELLA M. BASALE EPITELIALE Forma non familiare, non progressiva, spesso sottodiagnosticata. 4 espressioni cliniche: accumuli puntiformi, cisti, a carta geografica, a colpo d’unghia Per lo più asintomatica. Nel 10% dei casi lesioni corneali ricorrenti (fotofobia). REIS-BÜCKLERS Autosomica dominante Deposito progressivo di bande collagene sulla m. di Bowmann Fotofobia per erosioni corneali ricorrenti Terapia: PTK, trapianto DEGENERAZIONE EPITELIALE DI MESMMANN Autosomica dominante Degenerazione cistica epiteliale con anormalità della membrana basale Paucosintomatica SCHNYDER Autosomica dominante Deposito progressivo sub epiteliale cristallinico di ac. grassi neutri e colesterolo Haze = PTK, trapianto DEG. A LATTICE Forme di varia gravità Progressivi depositi amiloidi stromali Haze bollosa: esito in trapianto corneale GRANULARE autosomica dominante progressivi e confluenti depositi ialini amorfi Terapia : trapianto nel tipo I DISTROFIA ENDOTELIALE DI FUCHS Più frequente nelle donne Associata spesso a glaucoma cronico primario e cataratta Autosomica dominante, progressiva Haze bollosa = trapianto DISTROFIA ENDOTELIALE DI FUCHS Livello lesionale: endoteliale Clinica: Stadio iniziale: cornea guttata, con piccole escrescenze sulla superficie endoteliale asintomatico Stadio sintomatico: edema stromale, poi epiteliale disturbi visivi ingravescenti, cheratopatia bollosa dolore da rottura delle bolle Stadio terminale: cicatrizzazione terminale Complicanze: erosioni, ulcere, vascolarizzazione e degenerazione calcarea. Terapia: cheratoplastica perforante Lesioni guttate a livello endoteliale CHERATOENDOTELIOSI O DISTROFIA POLIMORFA POSTERIORE ENDOTELIALE È la vera distrofia presente sempre alla nascita Bilaterale Depositi sull’endotelio vescicolari o a carta geografica, o a banda Più o meno associata a glaucoma Per lo più asintomatico DEGENERAZIONI E DISTROFIE CORNEALI Cheratocono: Ectasia conica della cornea con assottigliamento dello stroma centrale correlata ad anomalia metabolica genetica dei cheratociti relativamente all’assemblaggio di glicoproteine. Bilaterale in oltre il 90% Pachimetria corneale Segno di Munson Sfiancamento centrale della cornea CHERATOPATIA A BANDELLETTA Opacizzazione corneale di origine calcica a livello della fessura interpalpebrale Etiologia : malattie generali con alterazioni del metabolismo fosfocalcico, tossici locali, infiammazioni, raramente ereditaria Inizia al limbo nasale e temporale a livello della fessura interpalpebrale poi si sviluppa nell’arco di molti mesi Non scompare mai spontaneamente e può riformarsi dopo la sua ablazione Depositi calcici a “quarto di luna” paralleli al limbo, raramente progrediscono verso il centro , possibile il raggiungimento della congiuntiva Istologicamente consiste in un deposito calcico extracellulare sulla membrana basale epiteliale della Bowmann e dello stroma anteriore GERONTOXON Opacità corneale periferica più frequente Deposito lipidico anulare parallelo al limbo, separato da esso da una zona chiara Generalmente bilaterale e simmetrico Di colore giallo-biancastro Bordi netti sul versante esterno, più sfumati su quello interno Inizia a livello della Bowman, superiormente e inferiormente per poi estendersi circonferenzialmente a 360° L’incidenza cresce con l’età ( 15% a 40 aa; 75% a 70 aa) L’acuità visiva è conservata perché non interessa la visione centrale Dopo i 50 aa considerato fisiologico, nei pz con meno di 50 aa si associa a Iperlipidemia primitiva e malattie cardio-vascolari ateromatose CHERATOPATIA DI LABRADOR La forma primitiva si distingue per l’assenza di precedenti oculari Consiste in una fine opacità simile a goccioline giallastre e dorate a livello della fessura interpalpebrale Generalmente l’esordio è al limbo nasale e temporale , ma le lesioni possono coinvolgere anche la congiuntiva A uno stadio più tardivo le goccioline formano una banda attraversante il centro della cornea con zone chiare e possibili rilievi La forma secondaria si caratterizza per l’ esistenza di cicatrice congiuntivale e stromale, assottigliamento stromale, neovascolarizzazione, testimoni di una malattia cronica come tracoma o congiuntivite batterica ricorrente In questo caso le lesioni a goccioline si formano su lesioni pre-esistenti La forma familiare non si distingue dalla primitiva tranne che per il ritrovamento in altri membri della stessa famiglia CHERATOPATIA DI LABRADOR Causa esatta sconosciuta, associata a condizioni climatiche estreme come i climi antartico e desertico Le persone che lavorano in ambiente esterno hanno un rischio maggiore di insorgenza Le radiazioni UV ,soprattutto riflesse, hanno un ruolo importante Può insorgere su pinguecola Istologicamente consiste in depositi globulari a livello della Bowman e dello stroma anteriore Non si conosce ancora la natura del contenuto delle goccioline Il trattamento di scelta è la fotocheratectomia terapeutica, cheratoplastica lamellare o perforante in un secondo tempo Prevenzione : protezione dalle aggressioni climatiche SOVRACCARICO LIPIDICO Deposito lipidico a livello stromale più frequentemente associato a NV corneale Talvolta dovuto a turbe del metabolismo lipidico Nelle NV appare come deposito biancogiallastro di lipidi intorno al tratto terminale dei neovasi I depositi sono densi e hanno margini spigolosi o lanuginosi Profondità dei depositi variabile, può invadere tutta la cornea La cornea superficiale rimane sempre liscia Spesso l’estensione e la densità dei depositi provocano una compromissione della acuità visiva SOVRACCARICO METALLICO L’anello di Kaiser-Fleischer si ritrova principalmente nella malattia di Wilson, ma può occorrere anche nel corso di altre epatopatie non wilsoniane E’ un deposito di rame periferico a livello della Descemet di colore arancione, bruno, verdastro o grigio a topografia superiore e inferiore che in seguito diviene circonferenziale Depositi corneali rameici possono essere osservati anche in presenza di corpo estraneo intraoculare La linea di Hudson-Stahli si ritrova dopo traumi e negli anziani. E’ costituita da emosiderina, di colore bruno, è situata orizzontalmente a livello delle cellule basali dell’epitelio più spesso alla giunzione del terzo inferiore e il terzo medio della cornea. Non si estende al limbo SOVRACCARICO FARMACOLOGICO L’ Epinefrina può indurre formazione di depositi congiuntivali di colore bruno I Sali d’Argento usati in forma topica possono provocare in cronico una argirosi che consiste in depositi blu-grigi a livello Descemet e congiuntiva come anche Sali di Mercurio e la Pilocarpina I Chinolonici inducono depositi cristalloidi sull’epitelio. La cornea verticillata è una forma clinica epiteliale caratterizzata da depositi intraepiteliali di particelle lipidiche a forma di vortice al terzo inferiore corneale. I farmaci maggiormente sospetti sono: amiodarone, clorochina, clorpromazina, indometacina, naprossene ma anche molti altri INFEZIONI Citomegalovirus intrauterino Incidenza infezione= 0.5-2.5% 10-20% mostra anomalie congenite Manifestazioni oculari: corioretinite (retinite acuta con aree di pigmentazione e atrofia), atrofia ottica, microftalmo, cataratta, cheratite. INFEZIONI Rosolia Glaucoma: 10%, più frequente negli occhi microftalmici Raramente neovascolarizzazione subretinica Cornea normale o lievemente opaca Cataratta: 20% dei bambini con sindrome rubeolica congenita Retinopatia rubeolica: 40% è bilaterale, con alterazioni EPR, più frequenti al polo posteriore. INFEZIONI Sifilide intrauterina Sifilide congenita: dopo 16ª sett. gestazione. Manifestazioni oculari: - corioretinite: aree periferiche pigmentate (pseudoretinite pigmentosa) - cheratite interstiziale (edema corneale e conseguente cicatrizzazione corneale con vasi fantasma) - uveite anteriore Diagnosi sierologica Trattamento: penicillina Anomalie di trasparenza: dermoidi •Si presume che la sua formazione sia dovuta al sequestro di elementi epidermici e connettivali durante l’embriogenesi. •Rappresenta un inclusione dermica ectopica che si accompagna a volte ad un coloboma palpebrale. •Queste anomalie rientrano nel quadro di una sindrome del primo arco branchiale. Occasionalmente circonda il limbus (dermoide anulare) oppure è bilaterale e può costituire uno dei componenti della sindrome di Goldenhar. •Spesso abbiamo un bambino ipovedente Essere ipovedenti è un problema che supera la percezione visiva ma investe nel bambino tutto lo sviluppo gnosico e l’attività prassica per l’oculomotricità intenzionale e organizzata. Ne risulta inficiata la sintesi simbolica, base del linguaggio verbale e funzioni quali l’attenzione selettiva (selezione dello stimolo), quale la memoria a breve ed a lungo termine (informazione iconica) e, in definitiva, la possibilità di acquisire la percezione analitico-sintetica delle parti e del tutto. Quando avviare il bambino alla riabilitazione? Valutare il grado di compromissione funzionale e l’entità del residuo visivo è fondamentale per l’intervento riabilitativo, sempre comunque prescolare e personalizzato, basato su un confronto interdisciplinare FLOGOSI CORNEALE La cornea risponde agli stimoli flogogeni secondo 2 modalità: 1. Risposta stromale: edemizzazione per tentare di diluire l’agente flogogeno 2. Risposta limbare: infiltrazione dello stroma da parte di cellule infiammatorie richiamate dal circolo limbare Edema corneale: L’edema corneale consegue ad un’alterazione dei meccanismi che regolano l’equilibrio idrico della cornea. Edema stromale: da patologie delle cellule endoteliali aumento di spessore della cornea da accumulo di liquido nello spazio interfibrillare formazione di pliche della Descemet (cheratite striata) Edema epiteliale: consegue all’edema stromale in stretta correlazione con la pressione intraoculare accumulo di liquido prima intercellulare poi intracellulare riduzione dell’acuità visiva, aloni colorati intorno alle luci, dolore Complicanze dell’edema corneale: 1. perdita di trasparenza 2. alterazione dei processi metabolici, con accumulo di metabolici tossici 3. fragilizzazione delle barriere endoteliale ed epiteliale con accumulo di liquidi e penetrazione di microrganismi NEOVASCOLARIZZAZIONE La cornea fisiologicamente è completamente avascolare. Lo sviluppo di neovasi costituisce la risposta estrema a condizioni patologiche che turbano l’equilibrio corneale e rappresenta un tentativo si supplenza a deficit di nutrizione. Le anse neovascolari originano dai plessi episclerali e progrediscono verso il focolai di flogosi, favorite dall’edema. La neovascolarizzazione può regredire. Panno: infiltrato cellulo-vascolare che interessa la metà superiore della cornea, espandendosi a Superficiale “pioggia” dal limbus al centro corneale. (es. tracoma) Profonda ULCERE CORNEALI PATOGENESI E CLINICA Eziopatogenesi: Forme da miceti filamentosi (Aspergillus): tipiche delle zone rurali Forme da lieviti (Candida): tipiche degli immunodepressi Aspetti clinici: Infiltrato biancastro irregolare con proiezioni digitiformi e focolai satelliti Possibile ipopion Dolore, fotofobia, sensazione di corpo estraneo Terapia: Aspergillus chetoconazolo Candida flucitosina CHERATITI FUNGINE ULCERA DENTRITICA DALL’ULCERA ALLA CHERATITE Herpes simplex Eziopatogenesi e forme cliniche: Forme virali, legate alla presenza del virus Cheratocongiuntivite della prima infezione: virus localizzato nelle cellule epiteliali; comparsa di ulcere, classicamente dendritiche o di lesioni aspecifiche come la cheratopatia puntata superficiale. Si associano una congiuntivite follicolare acuta monolaterale, una linfadenopatia preauricolare e tipiche lesioni cutanee da HSV; decorso spontaneo verso la guarigione entro 15gg, frequenti le recidive. Sintomi: irritazione, fotofobia e dolore Cheratite erpetica recidivante: provocato dalla diffusione del virus dal serbatoio di latenza verso la periferia in presenza di eventi che riducono la risposta immunitaria cellulo-mediata; comparsa di cheratite dendritica o a carta geografica; decorso: guarigione o evoluzione verso la forma metaerpetica. • Cheratite stromale: il virus giunge allo stroma dall’epitelio, dal limbus o dall’endotelio: edema, infiltrazione e precoce neovascolarizzazione che portano a fenomeni cicatriziali. • Cheratoendotelite: il virus giunge all’endotelio dallo stroma, dal limbus o dall’endotelio: edema disciforme; evoluzione: restitutio ad integrum o cicatrizzazione. Forme metaerpetiche, virus assente ma presenza di alterazioni dell’innervazione corneale in seguito a ripetuti episodi di cheratite erpetica: manifestazioni ulcerative (ulcera metaerpetica) ed edematose (cheratopatia bollosa metaerpetica). Terapia: Acyclovir in pomata oftalmica Interferone (Colliri cortisonici nelle forme stromali o endoteliali) CHERATITE DA VIRUS VARICELLA-ZOSTER Eziopatogenesi: Il virus HZV, dopo la prima infezione, rimane latente in un ganglio sensitivo e da qui può riattivarsi e raggiungere il territorio innervato dal ramo sensitivo. Nel 10% vi è un’interessamento della branca oftalmica del trigemino e può colpire la cornea. Aspetti clinici: Rash cutaneo con formazione di pustole, papule e ulcere ricoperte da croste. La cheratite evolve attraverso 4 stadi: 1. Cheratite epiteliale puntata: nel 50% dei casi; multiple lesioni periferiche puntate. 2. Cheratite microdendritica: piccole lesioni dendritiche non ulcerate a forma stellata, periferiche. 3. Cheratite nummulare: fini infiltrati subepiteliali. 4. Cheratite disciforme: raramente, dopo 20gg dalla comparsa del rash. Complicanze: Cheratouveite, Grave nevralgia post-erpetica Terapia: Acyclovir per os e pomate antibiotico-steroidee per le lesioni cutanee. CHERATITI BATTERICHE Eziopatogenesi: di solito conseguono a lesioni corneali superficiali (sopratutto traumatiche) che minano l’integrità della barriera epiteliale. In questi casi: Agenti eziologici: Staphilococco Aureus, Streptococco epidermidis e Pseudomonas Aeruginosa; Agenti eziologici con epitelio integro: Neisseria Ghonorrea e Meningitidis e Corynebacterium Dyphteriae. Aspetti clinici: Infiltrato corneale bianco-giallastro che può evolvere in ascesso corneale. Edema stromale Iperemia congiuntivale Possibile cheratoipopion Terapia: Antibiotici in collirio e iniezioni sottocongiuntivali, colliri cicloplegici. CHERATITI NON INFETTIVE Cheratocongiuntivite secca Eziopatogenesi: Alterazioni quantitative e qualitative del film lacrimale da: Deficit della componente acquosa: atrofia primitiva della ghiandola lacrimale sindrome di Sjögren (xerostomia) Deficit della componente mucosa: avitaminosi A (xeroftalmia) esiti cicatriziali di gravi congiuntiviti Aspetti clinici: Notevole discrepanza fra la sintomatologia soggettiva, molto accentuata, e l’obbiettività, spesso del tutto negativa. Cheratopatia puntata superficiale Possibili ulcere corneali Iperemia congiuntivale Sintomi: sensazione di corpo estraneo e bruciore, possibili annebbiamenti visivi transitori Terapia: lacrime artificiali in colliri o pomate. Cheratopatia da lagoftalmo o da esposizione Lagoftalmo: incompleta chiusura delle palpebre. Si ha di conseguenza un’esposizione permanente della superficie oculare: secchezza Eziopatogenesi: Esoftalmo Paralisi del VII nervo cranico (lagoftalmo paralitico) Alterazioni cicatriziali palpebrali Stati cachettici Aspetti clinici: cheratite puntata superficiale ulcere corneali fenomeni di cheratinizzazione congiuntivali e corneali Terapia: lacrime artificiali, bendaggio dell’occhio durante la notte, talvolta tarsorrafia Cheratopatia neurotrofica Eziopatogenesi: in seguito a compromissione della branca oftalmica del trigemino, si verifica un’alterazione del trofismo corneale con: cheratopatia puntata superficiale ulcere processi cicatriziali neovascolarizzazione. Sintomatologia: nessun dolore per mancanza di innervazione sensitiva Terapia: lacrime artificiali, eventuale bendaggio temporaneo Cheratiti associate a collagenopatie Coinvolgimento corneale in corso di artrite reumatoide, lupus eritematoso sistemico, granulomatosi di Wegener e poliarterite nodosa. Aspetti clinici: infiltrati stromali periferici ulcere. Nei casi più gravi: assottigliamento corneale e neovascolarizzazione. Nelle collagenopatie autoimmuni: possibile presenza di vasculiti retiniche, scleriti e neuropatias ottica ischemica. Cheratite in corso di artrite reumatoide Terapia: terapia della malattia di base; localmente: colliri cortisonici. Cheratiti in corso di malattie dermatologiche Affezione cutanea più spesso associata a cheratite: acne rosacea. Interessamento corneale nel 5% dei casi; Neovascolarizzazione con possibile formazione di un panno; talora ulcere ed assottigliamenti periferici o centrali. Terapia: locale, con colliri steroidei e tetracicline per os. Ulcera di Mooren (Ulcus Rodens) Rara e grave cheratite bilaterale di probabile origine autoimmune, in cui compare un’ulcera periferica al limbus, senza soluzione di continuità fra sclera e cornea. L’ulcera presenta tipicamente un bordo sottominato e gradualmente si estende lungo la circonferenza corneale. Segue un processo riparativo con cicatrizzazione, neovascolarizzazione ed assottigliamento corneale. Terapia: cortisonici locali. Cheratite marginale Cheratite abbastanza comune; sopratutto in pz affetti da blefarocongiuntivite stafilococcica. Ulcera corneale tipicamente periferica. Terapia: steroidi topici USTIONI CORNEALI Le ustioni corneali di natura chimica e termica rappresentano il 12-19% dei traumi oculari. Nella maggioranza dei casi si tratta di danni da agenti chimici, con un rapporto di 1:4 tra sostanze acide e sostanze basiche. Quasi il 30% delle causticazioni corneali di natura chimica coinvolgono entrambi gli occhi. Danno sul lavoro 68,1% Danno in ambiente domestico 24,1% Danno non identificato 7,8% Ustione da alcali 58,1% Ustione da acidi 14,1% Ustione termica 16,2% Ustione da altri agenti 11,6% Reim M., Redbrake C.: Chemical and Thermal injuries of the eyes. Surgical and medical treatment based on clinical and pathophysiological findings; Archivos de la Sociedad Espanola de Oftalmologia, Febbraio 2001 Il danno da acidi caratteristicamente si autolimita. Questo accade principalmente per due ragioni: Le cellule epiteliali coagulate (necrosi coagulativa) creano una sorta di barriera alla penetrazione dell’agente lesivo, proteggendo lo stroma corneale e le strutture intraoculari Le proteine dello stroma hanno un effetto tampone sull’azione dell’acido fino a un pH di 4 ASPETTI CLINICI E’ importante ricordare come la penetrazione dell’agente lesivo sia correlata con le condizioni di salute dell’epitelio corneale: in presenza di epitelio corneale integro, infatti, si verifica una discreta protezione contro la penetrazione di acidi deboli o diluiti fino ad un pH di 2,5. In seguito all’ablazione dell’epitelio corneale, invece, si realizza un danno severo anche in presenza di pH decisamente superiore a 2,5. GRADO I GRADO II GRADO III GRADO IV Erosione Iperemia PROGNOSI BUONA Erosione Ischemia 1/3 Chemosi PROGNOSI BUONA Erosione Ischemia > ½ Chemosi Opacità corneale (nascosti dettagli iridei) PROGNOSI RISERVATA Ischemia profonda >3/4 Opacità corneale densa ( non visibili dettagli iridei) Ischemia Sclerale Atrofia e decolorazione iride Essudati fibrinosi Proliferazione Ampie ulcere Cataratta Glaucoma Cicatrizzazione PROGNOSI SCARSA GRADO I Ustione lieve caratterizzata da aree di iperemia e chemosi congiuntivale, con piccole emorragie sparse che possono confluire in emorragie sotto congiuntivali più ampie. L’epitelio può essere intatto modestamente edematoso, o presentare solo disepitelizzazioni superficiali. Lo stroma si mentiene trasparente o leggermente edematoso. La C.A. à di profondità normale, otticamente vuota o con scarsa cellularità, il cristallino è trasparente, non ci sono modificazioni della pressione intraoculare. GRADO II Le ustioni moderate si associano spesso a lesioni della cute perioculare. La congiuntiva è iperemica e chemotica, i capillari congiuntivali perilimbari ed episclerali appaiono trombosati in alcune zone che si presentano come aree sbiancate. La cornea si presenta con aree di disepitelizzazione ampia o quasi totalmente disepitelizzata. Lo stroma appare edematoso e opaco, ma permette ancora di apprezzare i dettagli dell’iride e del margine pupillare. La C.A. è spesso cellulata, e vi può essere un temporaneo aumento della I.O.P. GRADO III La congiuntiva appare iperemica e con aree ischemiche in più di metà della circonferenza oculare. L’opacità corneale è maggiore, rendendo difficoltosa l’esplorazione dei dettagli iridei e pupillari. Il cristallino all’inizio sembra trasparente, ma può opacizzarsi in seguito. In caso di ustione con acido cromico e nitrico, l’epitelio congiuntivale e corneale appare giallo o bruno, e si desquama nel giro di alcuni giorni lasciando la congiuntiva iperemica e chemotica e lo stroma corneale chiaro. GRADO IV Le ustioni severe coinvolgono la cute della fronte, delle palpebre, del naso e delle guance. A livello palpebrale, il tessuto infiammatorio porta alla formazione di cicatrici retraenti che inducono in un secondo momento simblefaron e restrizione dei movimenti oculari. La formazione di cicatrici nella congiuntiva e nei tarsi e la distruzione dei margini palpebrali portano a un danno secondario su un epitelio già fragilizzato, principalmente per due ragioni: durante l’ammiccamento le cicatrici tarsali sfregano l’epitelio le cicatrici del margine palpebrale modificani la direzione delle ciglia inducendo trichiasi GRADO IV La congiuntiva appare iperemica con estese aree ischemiche. La cornea è marcatamente ispessita e totalmente opaca, per cui non si distinguono i dettagli dell’iride, della pupilla e della lente. Compare una marcata reazione iridociclitica con essudazione fibrinosa ed aumento della I.O.P. Nei casi più severi si ha anestesia corneale completa con pallore perilimbare e iridociclite florida. Nell’ustione da acido solfurico, il danno diretto ai nervi corneali porta ad anestesia, mentre nelle ustioni da acido fluoridrico gli ioni fluoruro combinandosi con calcio e magnesio in complessi insolubili immobilizzano il calcio, causando la stimolazione dei nervi corneali. USTIONI CHIMICHE DA ALCALI Le ustioni da alcali ( soda, ammoniaca, calce, cemento) sono le più pericolose perché diffondono nell’occhio in profondità ( cataratta, glaucoma, uveiti) Possono condurre a fusione del globo, retrazione palpebrale, congiuntivite fibrosante La prognosi corneale dipende dall’interessamento del limbo Lo sbiancamento della congiuntiva è un fattore prognostico negativo Il primo trattamento è un lavaggio abbondante con irrigazione, tp medica con Vitamina C, tetracicline, corticosteroidi topici e antibiotici topici per prevenire un’infezione, tp chirurgica In questa fase la terapia, ferma restando la necessità di un’adeguata perfusione e del sostegno farmacologico con inibitori della collagenasi (N-acetil-cisteina 10-20% ogni ora, EDTA, siero omologo), è principalmente di tipo chirurgico e prevede: LISI DELLE ADERENZE del tessuto necrotico congiuntivale e sottocongiuntivale con IMPIANTO DI GUSCI SCLERALI per mantenere l’architettura dei fornici NECROSECTOMIA, rivolta ad asportare il tessuto ischemico che costituisce un continuo stimolo infiammatorio. La meticolosa escissione del tessuto necrotico risparmia la sclera e la cornea.
Scaricare