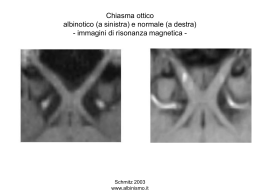
IL DEFICIT VISIVO PROGRESSIVO: ORIENTAMENTO AD UNA DIAGNOSI DIFFERENZIALE COMPLESSA Livello intermedio Direttore: S. Bianchi Marzoli Istruttori: Stefania Bianchi Marzoli, Arturo Carta, , Paola Ciasca, Barbara Giambene, Gemma Tremolada Introduzione Nella pratica clinica l’oftalmologo spesso deve gestire problematiche di difficile inquadramento clinico-diagnostico poiché molte condizioni patologiche presentano aspetti clinici simili. Pertano è fondamentale impostare un corretto iter diagnostico che consenta in primo luogo di eseguire un accurato esame neuroftalmologico e successivamente di mirare la richiesta di esami strumentali specifici, spesso molto indaginosi sia per tempo che costo. Il corso ha come scopo quello di fornire gli elementi clinici di base per potere formulare delle ipotesi diagnostiche e per la differenziazione tra le diverse patologie di pertinenza neuroftalmologica e retinica, responsabili di deficit visivo progressivo, attraverso l’ illustrazione di vari quadri clinici. Neuropatie ottiche compressive o glaucoma? Esistono forme di neuropatia ottica secondarie alla presenza di processi occupanti spazio che comprimono e inducono sofferenza delle fibre del nervo ottico. Sono per lo più forme tumorali, che possono svilupparsi dagli elementi stessi del nervo ottico oppure da strutture ad esso adiacenti. Tuttavia esistono anche forme secondarie ad anomalie vascolari o a fenomeni infiltrativi cronici di origine infiammatoria. Clinicamente si caratterizza da un progressivo deficit della funzione visiva, con calo dell'acuità visiva, deficit della percezione dei colori (discromatopsia) e pattern perimetrici spesso patognomonici di lesione posteriore intracranica quando estesi con rispetto del meridiano verticle. Possono associarsi deficit della motilità oculare estrinseca e, quando la lesione è intraorbitaria, esoftalmo e segni di congestione orbitaria. L'aspetto oftalmoscopico del disco ottico può essere diverso a seconda della sede della compressione del nervo; qualora questa sia anteriore si può sviluppare edema del disco ottico, mentre se si tratta di una compressione posteriore, il nervo ottico mostrerà, almeno nelle fasi iniziali, un aspetto normale. Nelle fasi tardive, indipendentemente dalla sede della lesione, il nervo apparirà pallido, fino all'atrofia diffusa, con spesso associato un aumento dell’ escavazione. A fronte di un sospetto clinico l'esame clinico prevede un accurata raccolta anamnestica e un accurato esame obiettivo neuroftalmologico completo di diagnostica strumentale quale la perimetria computerizzata (SAP) e l’ esame OCT RNFL e GCC (SD-OCT); da eseguire in seguito uno studio neuroradiologico mediante risonanza magnetica con Gadolinio mirata alla valutazione del nervo ottico in tutto il suo decorso, intraorbitario e intracranico. La TC può essere considerata un esame complementare alla RM, in grado di fornire informazioni aggiuntive e a volte dirimente nei casi di dubbio diagnostico, in grado di identificare la presenza di calcificazioni periottiche tipiche in caso di meningioma del nervo ottico; recentemente anche la PET si è dimostrata indicata nello studio dei meningiomi della guaina del nervo ottico. 1 La prognosi oltre ad essere funzione della causa determinante la neuropatia, è strettamente correlata alla durata e all'entità della compressione. I tumori primitivi del nervo ottico sono rappresentati dai gliomi e dai meningiomi. Le forme secondarie sono quelle che coinvolgono il nervo ottico provenendo da localizzazioni adiacenti e comprendono i glioblastomi, i meningiomi di origine intracranica, i retinoblastomi e le neoplasie metastatiche. I gliomi del nervo ottico costituiscono la più frequente causa di neuropatia ottica compressiva nel bambino e sono spesso associati a neurofibromatosi di tipo I; determinano riduzione della funzione visiva ed esoftalmo, mostrano lenta progressione e impongono trattamento chirurgico quando hanno tendenza allo sviluppo verso il canale ottico e il distretto intracranico. I meningiomi del nervo ottico originano dal rivestimento meningeo del nervo; si manifestano tipicamente con deficit della funzione visiva, atrofia ottica e shunt ottico-ciliari (vasi collaterali che drenano il sangue venoso dalla retina alla coroide). Come per i gliomi, il trattamento chirurgico è indicato solo quando mostrano tendenza alla crescita intracanalicolare o intracranica; allo stato attuale una valida alternativa è costituita dal trattamento radiante mediante radioterapia stereotassica frazionata. Anche le neoplasie a partenza della regione sellare (adenomi ipofisari, meningiomi del tubercolo sellare) possono determinare compressione a livello del tratto intracranico del nervo ottico. Accanto alle forme tumorali, vanno ricordate le forme vascolari, in particolare gli aneurismi (più frequenti quelli della arteria cerebrale anteriore, comunicante anteriore ed oftalmica) e le malformazioni vascolari del nervo ottico (emangiomi capillari, cavernosi e racemosi). Da menzionare la neuropatia ottica compressiva secondaria a conflitto neurovascolare il cui sospetto diagnostico è confermato mediante imaging neuroradiologico di RM. Anche l'orbitopatia tiroidea può determinare, nelle sue fasi più avanzate, una neuropatia ottica compressiva secondaria all'ispessimento dei muscoli extraoculari a livello dell'apice orbitario. La neuropatia ottica glaucomatosa tipicamente associata ad ipertensione intraoculare o le forme di glaucoma a tensione normale possono entrare in diagnosi differenziale sia per le modalità di presentazione clinica che decorso. Da ricordare anche la neuropatia ottica vascolare cronica spesso associata ad un condizione di ipoperfusione a carico delle strutture nervose del nervo ottico che osserviamo nei pazienti con una condizione clinicamente dimostrata, mediante esame Holter pressorio, di una brusca ipotensione notturna. Neuropatie ottiche ereditarie Le neuropatie ottiche eredodegenerative comprendono un gruppo eterogeneo di atrofie ottiche su base ereditaria nelle quali è costantemente presente un marcato deficit funzionale del nervo ottico. Le modalità di trasmissione sono diverse. Ricordiamo qui di seguito le principali. Neuropatia ottica autosomica dominante (ADOA). E’ la forma più comune; si caratterizza per un deficit visivo che insorge nella prima infanzia e mostra un carattere lentamente progressivo, con stabilizzazione del quadro intorno ai 10 anni. Sono tuttavia possibili casi ad esordio tardivo dopo i 40 anni. La compromissione visiva è bilaterale, anche se può essere asimmetrica, con scotoma centrale e deficit della percezione dei colori (discromatopsia). L'esame del fondo oculare mostra un pallore del disco ottico ed aumento dell'escavazione tipicamente temporale, con assottigliamento dello strato delle fibre nervose specie a carico del fascio papillo-maculare. L’esame OCT RNFL e GCC mostra quasi sempre un precocissimo e selettivo danno a carico del complesso ganglionare retinico. 2 Solitamente questa forma di neuropatia eredodegenerativa è isolata e i pazienti godono di buona salute generale; tuttavia sono stati segnalati casi associati a ritardo mentale, sordità, polineuropatia periferica. Nella maggior parte dei casi è secondaria alla mutazione a carico del gene OPA1 che codifica per una proteina della membrana mitocondriale interna. Neuropatia ottica autosomica recessiva Si tratta di un’ affezione estremamente rara, che si manifesta alla nascita (congenita) oppure nei primi anni di vita (infantile) con grave riduzione bilaterale dell'acuità visiva, fino alla cecità, ed è associata quasi sempre a nistagmo. L'esame del fondo oculare mostra una marcata atrofia ottica associata ad accentuazione dell’ escavazione del nervo ottico. A fronte di un sospetto clinico, è sempre indicato eseguire uno studio neuroradiologico mediante risonanza magnetica, al fine di escludere lesioni compressive intracraniche. Esistono forme associate a deficit neurologici importanti (atassie ereditarie) o quadri clinici complessi (diabete insipido centrale, diabete mellito, sordità, ritardo mentale come nella sindrome di Wolfram). Neuropatia ottica ereditaria di Leber (LHON). Si manifesta in giovani adulti, (ma possibile a qualsiasi età) prevalentemente di sesso maschile (può manifestarsi anche in soggetti di sesso femminile); è determinata da mutazioni puntiformi a carico del DNA mitocondriale trasmesse per via maternolineare. L'entità del deficit visivo sembra essere correlata alla percentuale di mitocondri che presentano la mutazione e alla eventuale presenza di fattori epigenetici scatenanti (abuso di alcool o fumo) e/o alla prresenza di deficit metabolici (Vit B12 e/o Folati). La sintomatologia è caratterizzata da riduzione dell'acuità visiva acuta o subacuta, monolaterale o bilaterale, rapidamente progressiva; se si presenta monolateralmente, il coinvolgimento dell'occhio controlaterale può verificarsi in un intervallo variabile, ma solitamente da qualche settimana a qualche mese. Il campo visivo mostra un profondo scotoma centrale. L'esame oftalmoscopico può mostrare in fase acuta un nervo ottico iperemico a margini sfumati (pseudoedema) con microteleangectasie (dilatazioni e tortuosità) dei vasi paripapillari; nelle fasi croniche appare evidente una atrofia settoriale a carico del settore temporale del disco ottico ed una perdita delle fibre nervose del fascio papillomaculare. L’esame OCT RNFL e GCC mostra quasi sempre un precocissimo e selettivo danno a carico del complesso ganglionare retinico. Il sospetto diagnostico può essere confermato dallo studio del DNA mitocondriale: in particolare circa il 90% dei casi di LHON è secondario ad una della tre mutazioni del DNA mitocondriale geneticamente determinate, 3460G>A, 11778G>A, 14484T>C che coinvolgono il complesso della catena respiratoria mitocondriale. Si tratta solitamente di una forma isolata, tuttavia in alcuni casi si può associare a coinvolgimento del sistema nervoso centrale (Leber plus) . La prognosi è variabile, potendosi anche verificare un miglioramento spontaneo. E' importante evitare il consumo di alcool e di fumo e associare una terapia di supporto con Vit B 12 e Folati. La terapia medica con Idebenone si è dimostrata una potenziale strategia terapeutica in grado di migliorare l’ entità del deficit visivo in termini di recupero dell’ AV nel 45% dei pazienti entro sei mesi dall’inizio della stessa; il trattamento si è inoltre dimostrato privo di effetti collaterali e ben tollerato. Distrofie retiniche occulte Le retinopatie centrali o generalizzate hanno caratteristiche cliniche che spesso possono essere confuse o entrare in diagnosi differenziale con le neuropatie ottiche. Questo soprattutto quando si considerano le forme di retinopatia occulta, per definizione difficilmente diagnosticabili alla sola esplorazione del fundus, poiché la retina può apparire di aspetto normale. 3 Presentazione comune alle due patologie è proprio un calo progressivo della funzione visiva che presenta caratteristiche confondenti: riduzione dell’acuità visiva centrale, discromatopsia, scotoma centrale, fotofobia, difetti perimetrici sovrapponibili, nonostante esistano segni clinici caratteristici che aiutano a differenziarle o quanto meno indirizzano il sospetto diagnostico. Nella fase diagnostica gli strumenti che possono aiutare un corretto orientamento sono sicuramente una attenta anamnesi ed un esame neuroftalmologico completo, ma la diagnosi di retinopatia non può prescindere da indagini strumentali fondamentali quali: 1) Esami elettrofisiologici (ERG full field, PERG, PEV): sono in grado di dimostrare l’alterata risposta dei fotorecettori, discriminando tra disfunzione che può interessare esclusivamente i bastoncelli, i coni od entrambi. Nel sospetto di retinopatia centrale occulta che quindi coinvolge prevalentemente il sistema dei coni, tramite l’elettroretinogramma multifocale (ERGmf) è inoltre possibile individuare l’entità del danno e localizzare topograficamente i settori maculari coinvolti 2) analisi SD-OCT: fornisce informazioni ultrastrutturali della retina e dei suoi segmenti ( strati retinici interni e strati retinici interni) attualmente con una alta risoluzione, fondamentali nella fase di diagnosi di retinopatia, e soprattutto di ausilio in corso di diagnosi differenziale. Sono infatti noti e descritti pattern tomografici tipici per diverse distrofie retiniche centrali e generalizzate. 3) Analisi in Autofluorescenza e Infrarosso: le immagini della retina ottenute tramite questa indagine forniscono informazioni di ordine metabolico che riguardano l’epitelio pigmentato retinico e quindi le strutture fotorecettoriali sovrastanti importanti non solo per il valore diagnostico ma anche prognostico in corso di retinopatia. Le retinopatie, soprattutto quelle occulte, esordiscono in modo non omogeneo manifestandosi con difetti strutturali anche in presenza di una funzione retinica normale, o con un difetto funzionale anche in assenza di danno ultrastrutturale. Questo rende ragione della necessità di effettuare tutte le indagini strumentali descritte per un corretto e precoce inquadramento. L’insieme delle informazioni cliniche/strumentali ci permettono infine di orientarci anche nella determinazione della possibile patogenesi. Le retinopatie, e soprattutto quelle definite ‘occulte’ riconoscono genesi diverse tra le quali citiamo distrofie retiniche centrali/generalizzate 1. geneticamente codificate: retinopatie di carattere eredo familiare, per alcune delle quali sono note mutazioni genetiche specifiche 2. di natura autoimmune: processi degenerativi della retina innescati dalla presenza di Ab antiretina 3. paraneoplastiche: retinopatie secondarie alla presenza di neoplasie tra le quali il melanoma ( MAR: melanoma-associated retinopathy) o cancro che può interessare diversi organi (CAR: cancer associated retinopathy) Spesso il processo di diagnosi diventa quindi multidisciplinare prevedendo la collaborazione con altri specialisti ( genetista, internista, oncologo…) per raggiungere una definizione. Deficit di origine non organica La diagnosi di questa entità clinica richiede un’ accuratezza di indagini atte ad escludere la natura organica del disturbo visivo. Spesso alla base di questa patologia esiste un tratto psicologico patologico o, in altri casi, aspetti legali spesso secondari a traumi. Si tratta di una condizione patologica in cui il dato soggettivo inerente i sintomi visivi lamentati dal paziente non trova nessun corrispettivo con il dato oggettivo inerente sia l’esame obiettivo neuroftalmologico che la diagnostica strumentale. L’esaminatore spesso denota un particolare 4 atteggiamento del paziente nel corso della visita, così come utilizza strategie di esame utili a smascherarlo. Nella maggior parte dei casi il paziente lamenta una perdita della visione, intesa sia come ridotta acuità visiva che come perdita del campo visivo mono o bilaterale. Non esiste un pattern perimetrico tipico in quanto questa categoria di pazienti è in grado di simulare difetti del campo visivo assolutamente variabili, anche se quello maggiormente riprodotto, nella pratica clinica, risulta essere il restringimento concentrico o a spirale delle isoptere ed il restringimento a quadrifoglio; la peculiarità consiste nella scarsa ripetibilità dell’esame. Proprio per questa tipicità di pattern perimetrico questa patologia può entrare in diagnosi differenziale con varie forme di retinopatia sia eredo-degenerativa che paraneoplastica (CAR). Diversi termini clinici sono stati utilizzati, in passato, per classificare questa entità patologica quali ambliopia psicogena, ambliopia isterica o cecità isterica. L’iter clinico diagnostico deve necessariamente escludere tutte le cause di patologia e quindi deve essere completo di diagnostica strumentale quale perimetria computerizzata, OCT (RNFL e GCC), esami elettrofisiologici (PEV, PERG ed ERG) e diagnostica neuroradiologica. Il percorso diagnostico e terapeutico è multidisciplinare e richiede spesso l’integrazione del quadro obiettivo neuroftalmologico con una valutazione psicologica e/o psichiatrica di supporto. Particolare attenzione va dedicata all’approccio terapeutico-psicologico nei confronti di questa categoria di pazienti. Una volta ipotizzata e confermata la natura del disturbo visivo lamentato dal paziente il clinico deve avere la massima attenzione a rassicurare il paziente sulla normalità dell’ esame obiettivo, sulla negatività delle indagini eseguite e sulla reversibilità e temporaneità del quadro clinico patologico. Spesso il paziente viene indirizzato ad una valutazione psicologica specialistica di supporto che mira ad identificare la causa scatenante il disturbo visivo. Deficit di origine chiasmatica e retro chiasmatica Le lesioni primitive e secondarie del chiasma determinano il pattern perimetrico tipico bitemporale. Le patologie a carico delle vie ottiche retro chiasmatiche sono spesso caratterizzate da pattern perimetrici patognomonici ed in alcuni casi si associano ad altri sintomi neurologici. Il chiasma ottico è una struttura a localizzazione intracranica che nasce dall'unione dei due nervi ottici e dà origine posteriormente ai tratti ottici; è alloggiato al di sopra della sella turcica dell'osso sfenoide, che contiene il corpo della ghiandola ipofisaria, ed è in stretto rapporto con i vasi del circolo di Willis. La disposizione delle fibre nervose del chiasma è tale da spiegare i difetti perimetrici secondari a lesioni di questa struttura, siano esse primitive o secondarie. All'interno del chiasma ottico si uniscono le fibre visive che provengono da ciascun nervo ottico; le fibre temporali hanno un decorso diretto e si portano al tratto ottico dello stesso lato, mentre le fibre nasali decussano e si portano al tratto ottico controlaterale. La manifestazione più caratteristica di danno del chiasma ottico è un difetto del campo visivo bilaterale dell'emicampo temporale che corrisponde a sofferenza delle fibre nasali crociate. La funzione del chiasma ottico può essere alterata (sindrome chiasmatica) dalla presenza di processi patologici primitivamente a carico del chiasma stesso, come neoplasie (gliomi) o lesioni infiammatorie-demielinizzanti (sarcoidosi , sclerosi multipla), oppure che interessano le strutture endocraniche adiacenti, come neoplasie endo e perisellari (adenoma ipofisario, craniofaringioma, mengioma dello sfenoide), malformazioni arterovenose (aneurismi del circolo di Willis) o processi 5 flogistici meningei della base. Gli adenomi ipofisari sono la causa più frequente di sindrome chiasmatica. Nella sindrome chiasmatica in fase iniziale il deficit campimetrico è bilaterale, ma spesso asimmetrico, e corrisponde ad una qudrantanopsia o emianopsia bitemporale. L'acuità visiva è solitamente normale, ma può andare incontro ad una compromissione di entità variabile al persistere della noxa patogena. Può essere presente diplopia binoculare da paralisi dei nervi oculomotori III, IV, VI, secondaria a compressione o invasione del seno cavernoso da parte della lesione. E' possibile anche la comparsa di nevralgia trigeminale. Altri sintomi possono essere la cefalea e, nei casi di adenoma ipofisario secernente, le disfunzioni endocrine variabili a seconda dell'ormone prodotto. Il sospetto diagnostico si fonda sul riscontro di pattern perimetrici patognomonici; l'aspetto oftalmoscopico è spesso normale, potendo mostrare solo nelle fasi tardive un pallore settoriale o diffuso della testa del nervo ottico (atrofia ottica). La diagnosi eziologia richiede uno studio neuroradiologico mediante RM mirata allo studio della regione sellare. La terapia è differenziata a seconda della causa determinante la sindrome chiasmatica. Il trattamento degli adenomi ipofisari è prevalentemente chirurgico, con approccio per via transfenoidale o per via transcranica nelle lesioni di maggiori dimensioni o con carattere infiltrativo. Le recidive o i residui possono essere trattati con radiochirurgia stereotassica frazionata e in alcuni casi anche con radioterapia convenzionale. La prognosi visiva è generalmente buona nei casi diagnosticati precocemente ed è funzione del danno causato dalla lesione, espressione dell'entità e della durata della compressione. Le vie ottiche retrochiasmatiche (tratto ottico, corpo genicolato laterale, radiazione ottica e corteccia occipitale) veicolano le fibre che originano dalla metà temporale della retina omolaterale e dalla metà nasale della retina controlaterale, in modo tale che ogni via contenga le informazioni provenienti dallo spazio visivo controlaterale. Questa distribuzione anatomica rende ragione del difetto bilaterale omonimo (dello stesso lato) che si verifica nei casi di danno a carico delle vie ottiche retrochiasmatiche. L'acuità visiva è solitamente conservata e l'aspetto del fondo oculare è normale. Poiché spesso viene riferita una sensazione di calo visivo monolaterale, a carico dell'occhio del quale è coinvolto l'emicampo temporale, solo un esame neuroftalmologico completo consente di rivelare la presenza di un difetto del campo visivo suggestivo di patologia del sistema nervoso centrale. Il danno delle vie ottiche retrochiasmatiche che si trovano nei lobi temporale e parietale è quasi sempre associato ad altri sintomi neurologici diversamente da quello che coinvolge la porzione più posteriore che si trova nel lobo occipitale esclusivamente dedicato alla visione. Le cause più frequenti di danno delle vie ottiche retrochiasmatiche sono di origine ischemica, neoplastica, congenita-malformativa, infiammatoria e infiltrativa. La corretta gestione del paziente in cui si sospetta un danno delle vie ottiche retrochiasmatiche, oltre ad una valutazione neurologica, richiede uno studio neuroradiologico completo mirato a valutare la presenza e la natura della lesione. La prognosi visiva è funzione del meccanismo patogenetico della lesione, dell'entità e della durata del danno, e dall'eventuale possibilità di eliminare la causa determinante. In base alla sede anatomica della lesione possiamo avere pattern perimetrici e quadri clinici peculiari: Lesioni del Tratto ottico I tratti ottici originano posteriormente al chiasma, si dirigono posteriormente e lateralmente, circondando i peduncoli cerebrali per terminare principalmente a livello del corpo genicolato laterale. Una lesione del tratto ottico isolata è piuttosto rara e solitamente è determinata da patologie che coinvolgono anche il chiasma ed eventualmente i nervi ottici; l'emianospia è in questo caso caratterizzata da una forte incongruità. Tipicamente si osserva una anomala reazione pupillare, chiamata difetto pupillare relativo afferente, che localizza la lesione a livello del tratto ottico 6 controlaterale all'occhio che mostra tale riflesso. Nelle fasi croniche si può riscontrare oftalmoscopicamente un pallore settoriale della papilla ottica, secondario alla degenerazione retrograda delle fibre nervose e cellule ganglionari. Lesioni del Corpo genicolato laterale Il corpo genicolato laterale si trova a livello talamico ed è la sede in cui le fibre visive che originano dalle cellule ganglionari retiniche entrano in sinapsi con i neuroni i cui assoni andranno a costituire le radiazioni ottiche. Una lesione isolata del corpo genicolato è rara. In questi casi non sono presenti alterazioni della funzionalità pupillare né modificazioni dell'aspetto del disco ottico. Il difetto perimetrico, omonimo, è a spicchi o a clessidra secondo la peculiare distribuzione retinotopica delle cellule neuronali. Lesioni della Radiazione ottica Dal corpo genicolato laterale origina la radiazione ottica, le cui fibre superiori (che veicolano informazioni relative all'emicampo inferiore) si dirigono direttamente verso la corteccia occipitale attraverso il lobo parietale, mentre le fibre inferiori (che portano informazioni provenienti dall'emicampo superiore) formano un'ansa intorno al sistema ventricolare a livello del lobo temporale (ansa di Mayer) prima di raggiungere la corteccia occipitale. Una lesione che interessa il lobo parietale provocherà di conseguenza un difetto omonimo dell'emicampo inferiore controlaterale (quadrantopsia omonima inferiore), mentre una lesione del lobo temporale, provocherà pertanto un difetto nel quadrante superiore (quadrantopsia omonima superiore). Spesso i sintomi visivi si associano a disturbi neurologici, quali emiplegia, afasia, agnosia, allucinazioni, epilessia. Lesioni della Corteccia visiva La corteccia visiva primaria è localizzata a livello del lobo occipitale, e comprende un'area visiva primaria (corteccia striata), situata superiormente e inferiormente alla scissura calcarina, e due aree associative (corteccia parastriata e peristriata). La maggior parte dell'area visiva primaria è deputata alla visione maculare (porzione posterolaterale); mentre al livello della corteccia che si trova lungo la fessura intraemisferica è rappresentato il campo visivo perifericoIl quadro clinico delle lesioni occipitali è caratterizzato dalla congruità del difetto omonimo del campo visivo, che può presentarsi anche sottoforma di scotomi e quandrantopsie. Tipico delle lesioni occipitali è il risparmio maculare, che si caratterizza per la conservazione dei 5-10° centrali, funzione della duplice vascolarizzazione del polo occipitale e della rappresentazione bilaterale della regione maculare. Quando la lesione si localizza bilateralmente nelle vie ottiche retrogenicolate o alla corteccia striata si parla di cecità corticale, che prevede il deficit completo delle funzioni visive, con riflessi pupillari conservati e normale aspetto del fondo oculare. Segni neurologici associati, indice di estensione alle aree visive adiacenti e alle vie di associazione, sono rappresentati da allucinazioni visive e anosognosia visiva (il paziente non riconosce la propria cecità). Le patologie responsabili di questo quadro clinico sono di natura vascolare ischemica, meno frequentemente di origine infettiva o tossica (da monossido di carbonio). In molti casi oltre alla RM encefalo può essere utilizzata anche la SPECT cerebrale in grado di rilevare fini alterazioni a carico del metabolismo tissutale cerebrale nelle patologie cerebrovascolari ed in quelle indotte da agenti tossici. 7 BIBLIOGRAFIA - Quantitative analysis of optic disc cupping in compressive optic neuropathy. Bianchi-Marzoli S, Rizzo JF, Brancato R, Lessell S. Ophthalmology 1995;102(3):436-40. - SPECT and PET Imaging of Meningiomas. Valatassiou V, Leondi A, Angelidis G, Psimadas D, Georgoulias P. The Scientific World Journal 2012; 412:1-11. Primary Optic Nerve Sheath Meningioma in Children. Lee HBH, Garrity JA, Cameron JD, Strianese D, BonovolontàG, Patrinely JR. Surv Ophtalmol 2008;53(6):543-55. - Diagnosis and Management of Optic Nerve Sheath Meningiomas. Shapey J, Sabin HI, Danesh Meyer HV, Kaye AH. J Clin Neurosci 2013; 20:1045-1056. - Treatment strategies for inherited optic neuropathies: past resent and future. Yu-Wai-Man P, Votruba M, Moore AT, Chinery PF. Eye 2014; 28(5):521-537. - Treatment of hereditary optic neuropathiese. Newman NJ. Nat Rev Neurol. 2012;8(10):545-56. - Pattern ERG: clinical overview, and some observations on associated fundus autofluorescence imaging in inherited maculopathy. Holder GE, Robson AG , Hogg CR, Kurz-Levin M, Lois N, Bird AC. Doc Ophthalmol 2003;106: 17–23. - Fundus autofluorescence in children and teenagers with hereditary retinal diseases Bettina Wabbels B, Demmler A, Paunescu K, Wegscheider E, Preising MN, Lorenz B. Graefe’s Arch Clin Exp Ophthalmol 2006;244:36–45. - Transition Zones between Healthy and Diseased Retina in Choroideremia and Stargardt Disease as Compared to Retinitis Pigmentosa. Lazow MA, Hood DC, Ramachandran R, Burke TR, Wang YZ, Greenstein VC, Birch DG. Invest Ophthalmol Vis Sci. 2011;52(13):9581-90. - Identification of functional visual field loss by automated static perimetry Acta Ophthalmol. 2014. ;92(8):805-9 - Diagnosis and Management of Functional Visual Deficits. Leavitt JA . Curr Treat Options Neurol. 2006 Jan;8(1):45-51 - Relationship between optical coherence tomography, pattern electroretinogram and automated perimetry in eyes with temporal hemianopia from chiasmal compression. Monteiro ML, Cunha LP, Costa-Cunha LV, Maia OO Jr, Oyamada MK. Invest Ophthalmol Vis Sci. 2009;50(8):3535-41. - Homonymous Ganglion Cell Layer Thinning After Isolated Occipital Lesion: Macular OCT Demonstrates Transsynaptic Retrograde Retinal Degeneration. Meier PG, Maeder P, Kardon RH, Borruat FX. J Neuroophthalmol 2014;0:1-5. 8
Scaricare