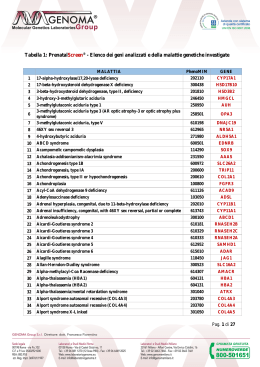
SINTOMI COMUNI E… MALATTIE RARE DANIELE DE BRASI GENETICA CLINICA UOC PEDIATRIA AORN SANTOBONO-PAUSILIPON CASO 1 Viviana, 2 anni scarsa crescita e ipertransaminasemia… Viviana, 2 anni Ricovero in DH di pediatria per scarsa crescita. • Nonni materni sordomuti • Gravidanza: normocondotta • P.S. 40w – Kg 3.100 • No asfissia perinatale • Screening udito OK • Storia di scarso accrescimento ponderale insorto già nel 1° anno di vita • Normale acquisizione tappe di SPM Viviana, 2 anni Ricovero in DH di pediatria per scarsa crescita. EO : P = Kg 11.500 (25°ct) A = cm 91 (90°ct) CC = 46 (10°ct) P/S 5°ct •Note di distrofia – fegato palpabile a 2 cm sulla xifo-ombelicale – soffio protosistolico 1-2/6 •Sviluppo cognitivo nei limiti con note comportamentali di iperattività •Facies caratterizzata da: fronte ampia, padiglioni auricolari sporgenti, ponte nasale pronunciato (somiglianza con il padre) •Telarca bilaterale. Nella norma: emocromo – indici flogosi – funzionalità renale – Ipertransaminasemia: AST 59 – ALT 76 – ALP 250 – gamma GT 48 Nella norma: bilirubinemia diretta ed indiretta – coagulazione - QPE Nella norma: screening infettivologico per virus epatitici maggiori e minori – Ig – elastasi fecale – anti-TTG – ceruloplasmina – cupremia e cupruria – alfa1 antitripsina – test del sudore CONSULENZA ENDOCRINOLOGICA: B2 PH1; statura al 75° ct nel bersaglio genetico, normale velocità di crescita: Telarca prematuro verosimilmente isolato. - RX MANO x ETA’OSSEA corrispondente all’EC - Ecografia pelvica con istero-annessometria strutture nei limiti per età - FSH 7.2 – LH 0.2 – E2 10 CONTROLLO: Telarca prematuro isolato. ECOGRAFIA ADDOME IN TOTO: - Fegato e vie biliari nei limiti - Reni e vie urinarie: doppio distretto renale a sx … SCARSA CRESCITA + IPERTRANSAMINASEMIA + ANOMALIE RENALI + FACIES PECULIARE … SCARSA CRESCITA + IPERTRANSAMINASEMIA + ANOMALIE RENALI + FACIES PECULIARE SI RICONOSCE CIO’ CHE SI CONOSCE… CHIEDIAMO: … CHIEDIAMO: ECOCARDIOGRAMMA - RX RACHIDE - VISITA OCULISTICA ECOCARDIOGRAMMA: stenosi di grado lieve di entrambi i rami della polmonare (gradiente max sn 30 mmHg, dx 25 mmHg), che appaiono di calibro ridotto RX RACHIDE: non significative lesioni a carico dei metameri esaminati (minimo cleft a carico di D12) VISITA OCULISTICA: FO con disco ottico a margini sfocati (OS>OD) leggermente sollevato sul piano, senza segni di compromissione vascolare. Macula in ordine in entrambi gli occhi. embriotoxon a sx., iride normocromica e normotrofica in entrambi gli occhi. CARIOTIPO: 46,XX Ripete INDICI FUNZIONE EPATICA 08.10 23.10 19.12 09.04 AST 59 94 112 65 ALT 76 106 116 101 ALP 250 317 269 273 GGT 48 72 79 74 … SCARSA CRESCITA + COLESTASI + ANOMALIE RENALI + FACIES PECULIARE + CARDIOPATIA CONGENITA + EMBRIOTOXON SINDROME DI … SINDROME DI ALAGILLE (OMIM #118450) Alagille-Watson syndrome, cholestasis with peripheral pulmonary stenosis, arteriohepatic dysplasia, syndromic hepatic ductular hypoplasia, EREDITARIETA’ DI TIPO AUTOSOMICO DOMINANTE – mutazioni di JAG1 (97%) – NOTCH2 (1-2%) EPATOPATIA COLESTATICA (95%) Spesso ad esordio neonatale (ipoplasia duttulare), con aumento indici epatici (gamma GT) e acidi biliari sierici. Possibile danno epatico progressivo. FACIES (80%) Aspetto del volto ‘a triangolo rovesciato’, occhi infossati, fronte ampia, padiglioni auricolari sporgenti, radice nasale depressa, mento appuntito RITARDO DI CRESCITA I pazienti con ALGS hanno di solito un basso peso alla nascita e una crescita staturale nei centili bassi della normalità e una magrezza sono frequenti. ANOMALIE SCHELETRICHE (40-85%) Difetto di fusione dell’arco vertebrale con immagine radiologica di vertebra ‘a farfalla’. ANOMALIE OCULARI (60-85%) Embriotoxon posteriore (persistenza dei residui embrionali dell’anello di Schwalbe alla giunzione irido-corneale). ANOMALIE RENALI (<40%) Agenesia, ipoplasia, cisti, ostruzioni uretero-pieliche, duplicazioni pieliche. ANOMALIE CARDIACHE (85-95%) Stenosi periferica di uno o più rami dell’arteria polmonare, difetti settali. ANOMALIE VASCOLARI Sono prevalentemente a carico del circolo vascolare cerebrale, ma anche dell’aorta e dell’arteria renale Criteri di diagnosi di ALGS Almeno tre dei cinque elementi più comunemente rappresentati: - malattia colestatica - facies - cardiopatia congenita - anomalie oculari - anomalie scheletriche PUZZLE COMPLETO!! MUTAZIONE de novo RM ENCEFALO CON SEQUENZE ANGIO: … Le sequenze angiografiche endocraniche eseguite in fase arteriosa e venosa mostrano: agenesia della carotide interna di dx e di A1 omolaterale. Il segmento A2 e l'arteria cerebrale media di dx sono rifornite rispettivamente dalla comunicante anteriore e da una prominente arteria comunicante posteriore omolaterale. Non si evidenziano alterazioni di alterazioni di calibro e decorso dei restanti principali rami arteriosi intracranici e dei seni venosi, né immagini da riferire a vasi neoformati e aneurismi. Ipoplasia del seno trasverso di destra. A2 A1 normale normale agenesia della carotide interna di dx e di A1 omolaterale agenesia della carotide interna di dx e di A1 omolaterale normale PRESENT CASE La prognosi della ALGS - Condizionata dalla severità delle anomalie cardiovascolari e dalla progressione della malattia epatica (sopravvivenza globale a 10 anni dalla diagnosi 68-80%, a 20 anni 62%-75%) - I pz ALGS hanno globale e significativa riduzione dello stato di benessere psicofisico (possibili altre patologie croniche dell’età pediatrica ad es. artrite cronica giovanile) - La mortalità è principalmente correlata alle malformazioni vascolari, all’eventuale presenza di cardiopatia congenita complessa e alla malattia epatobiliare. Trattamenti nella ALGS Malattia biliare. Farmaci coleretici (acido ursodesossicolico, rifampicina, colestiramina). L’indicazione al trapianto di fegato sono ritardo di crescita e prurito intrattabile. Cardiopatie congenite. Se complesse e sintomatiche trattamento chirurgico. Malnutrizione e ritardo di crescita. Apporto calorico superiore di almeno il 50% il fabbisogno per l’età, apporto di acidi grassi a catena media, adeguata e periodica somministrazione di vitamine liposolubili. ACKNOLEDGEMENTS Claudia Mandato U.O.C. Pediatria – Ospedale Santobono Angela Boccieri U.O.S. di Day Hospital – Ospedale Santobono CASO 2 Antonio, 2 anni 7 mesi ritardo dello sviluppo psicomotorio e convulsioni febbrili… Antonio, 2 anni 7 mesi Ricovero in pediatria per convulsione febbrile. • Familiarità per convulsioni febbrili • Gravidanza: normocondotta • P.C. 38 w emorragia intra-partum – Kg 2.910 • No asfissia perinatale • Screening udito OK • Storia di convulsioni febbrili (2 episodi pregressi) iniziate a 10 mesi • EEG: anomalie aspecifiche • TC cranio normale • Visita Oculistica (18 mesi): esotropia OS - ipopigmentazione FO • Ritardo acquisizione tappe di SPM • Normale crescita staturo-ponderale Antonio, 2 anni 7 mesi Ricovero in pediatria per convulsione febbrile. EO : P = Kg 29 (>>97°ct) A = cm 105 (>>97°ct) CC = 53.3 (>97°ct) P/S >>97°ct BMI = 26.36 (>>95°ct) Obesità severa – ritardo cognitivo – note dismorfiche Nella norma: routine ematochimica – indici flogosi – funzionalità d’organo – coagulazione – Ig – antivirali Consulenza cardiologica e ECG: nella norma Consulenza neurologica: RSPM, 3° episodio convulsivo in febbre, note dismorfiche: richiesti EEG e RM encefalo Consulenza genetica: Dott. Daniele De Brasi medico-chirurgo Specialista in Pediatria Specialista in Genetica Medica Consulenza endocrinologica: obesità di grado elevato, alta statura (superiore al bersaglio genetico), G1 – PH1, macrorchidia ai limiti (volume testicoli 3 ml); note dismorfiche (sindattiliabrachidattilia); problemi oculari; ritardo psicomotorio; richiesti: RX MANO E POLSO: età ossea di circa 2 anni TSH, FT4 FSH, LH, testosterone, DHEA-S Cortisolo basale Nella norma insulina basale e post-prandiale Consulenza oculistica: esotropia, epicanto, pupilla eccentrica, ipopigmentazione del fundus ECOGRAFIA ADDOME: nella norma EEG: AEC in sonno nella norma RM encefalo: normale Ritardo cognitivo + dismorfismi CHE TEST FARE ???? ARRAY-CGH ARRAY = matrice CGH = Comparative Genomic Hybridization CMA = CHROMOSOMAL MICROARRAY misurazione del rapporto tra le intensità di fluorescenza emesse da un DNA campione rispetto a un DNA di riferimento – ogni sonda permette di identificare le variazioni del numero di copie tra i 2 DNA 5-10 Mb Karyotype 300-600 bands BAC 100 Kb-1 Mb bacterial artificial chromosome CNVs SNPs 20-80 bp single-nucleotide polymorphism CITOGENETICA TRADIZIONALE SUBTELOMERI ARRAY-CGH 4p3.7% 7% 10.5% Test genetici e ritardo mentale Miller DT. Am J Hum Genet 2010 ANTONIO WARBURG MICRO SYNDROME CATEGORY SUBCATEGORY Inheritance Growth FEATURES Autosomal recessive Height Short stature Other Postnatal failure to thrive Head Microcephaly Ears Large ears Ears Large ears Eyes Microphthalmia Microcornea Congenital cataract Optic atrophy Ptosis and deep-set eyes Genitourinary External Genitalia (Male) Internal Genitalia (Male) Hypogenitalism Cryptorchidism Skeletal Feet Deformities of metatarsal bones (rare) Overlapping toes (rare) Neurologic Central Nervous System Mental retardation Hypotonia Hypoplasia/agenesis of the corpus callosum Cerebral/cerebellar malformations Head/Neck Molecular Basis Mutation in the RAB3 GTPase-activating protein subunit 1 gene (RAB3GAP1) WARBURG MICRO SYNDROME CATEGORY SUBCATEGORY Inheritance PRESENT PATIENT FEATURES Autosomal recessive Autosomal dominant Height Short stature Tall stature Other Postnatal failure to thrive Overgrowth Head Microcephaly Macrocephaly Ears Large ears Ears Large ears Low set ears Eyes Microphthalmia Microcornea Congenital cataract Optic atrophy Ptosis and deep-set eyes Exotropia Eccentric pupil Retinal hypopigmentation Genitourinary External Genitalia (Male) Internal Genitalia (Male) Hypogenitalism Cryptorchidism Scrotal hypoplasia Skeletal Feet Deformities of metatarsal bones (rare) Overlapping toes (rare) Brachy-syndactyly Neurologic Central Nervous System Mental retardation Hypotonia Hypoplasia/agenesis of the corpus callosum Cerebral/cerebellar malformations Mental retardation Hypotonia Mutation in the RAB3 GTPase-activating protein subunit 1 gene (RAB3GAP1) Duplication of 2q21.3 region, including RAB3GAP1 gene Growth Head/Neck Molecular Basis 5q35 CONTROFENOTIPO (reversed phenotype) DELETION (Sotos syndrome) DUPLICATION (Microduplication) - 7p11.2 DELETION (Williams-Beuren syndrome) DUPLICATION (Williams-Beuren region Duplication syndrome) - 17p11.2 DELETION (Smith-Magenis syndrome) DUPLICATION (Potocki-Lupski Syndrome) - 22q11.2 DELETION (CATCH 22/DiGeorge/Velocardiofacial syndrome) DUPLICATION (Microduplication) - … 2q21.3 DELETION (homozygous) (Warburg-MICRO) DUPLICATION (Microduplication) – PRESENT CASE + … REVERSE DYSMORPHOLOGY (Genotype First) … The classical practice of identification of the phenotype first and analysis of the genotype second is reversed. In this way, clusters of cases are first recognized by their overlapping copy number changes and subsequently the phenotypes of the individuals in the cluster are compared. This new approach permits the recognition of common clinical features that were initially too subtle or too variable (when seen in patients of different age, sex, and ethnicity) to enable a new syndrome to be identified solely on clinical grounds. This approach may be particularly valuable for the investigation of disorders with high locus heterogeneity, e.g., learning disability. … From: The American Journal of Human Genetics 84, 524–533, April 10, 2009, modified ACKNOLEDGEMENTS Rita Genesio A.F. Citogenetica e Diagnosi Prenatale – AOU “Federico II” Sintomi (e segni) comuni e malattie rare Messaggi conclusivi 1. Sintomi comuni e malattie (rare): dietro sintomi (e segni) comuni possono nascondersi condizioni rare: impariamo a pensarci (ma pensare sempre prima a malattie comuni, poi a forme rare di malattie comuni, poi a forme comuni di malattie rare). 2. Nuovo approccio clinico-diagnostico al bambino con sospetta condizione genetica: la REVERSE DYSMORPHOLOGY come “modello” di approccio diagnostico. 3. Malattie rare (ma non troppo): riconoscerle prima per curarle meglio.
Scaricare