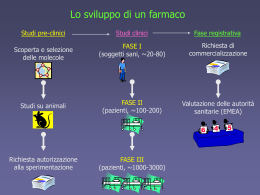
LE SPERIMENTAZIONI CLINICHE FASI della SPERIMENTAZIONE CLINICA FASE I: è la definizione dell’ambito di dosi singole ben tollerate nell’uomo con conseguente scelta del dosaggio da adottare nei successivi studi terapeutici FASE II: è il saggio di attività terapeutiche specifiche con la conseguente identificazione delle principali indicazioni di impiego. FASE III: è la stima dell’attività terapeutica nelle principali indicazioni in condizioni vicine il più possibile alla pratica clinica FASE IV: è la stima accurata del rapporto sicurezza/efficacia in condizioni di usuale utilizzazione medica del trattamento. FASI della SPERIMENTAZIONE CLINICA FASE I: è la definizione dell’ambito di dosi singole ben tollerate nell’uomo con conseguente scelta del dosaggio da adottare nei successivi studi terapeutici Si effettua generalmente su volontari sani Il numero di soggetti generalmente studiati è di 10-40 La scelta della dose iniziale, generalmente molto piccola, è guidata da studi farmacologici, farmacocinetici e tossicologici FASI della SPERIMENTAZIONE CLINICA FASE II: è il saggio di attività terapeutiche specifiche e della tossicità con la conseguente identificazione delle principali indicazioni di impiego. Si effettua su pazienti nell’ambito di studi non comparativi e comparativi di limitata numerosità e di breve durata Il numero di soggetti generalmente studiati è di 50-100 FASI della SPERIMENTAZIONE CLINICA FASE III: è la stima dell’attività terapeutica nelle principali indicazioni in condizioni vicine il più possibile alla pratica clinica Si effettua su pazienti nell’ambito di studi comparativi in confronto placebo o con farmaci già disponibili e considerati standard Permette di ottenere una conferma del rapporto sicurezza/efficacia Serve meno a valutare anche la tollerabilità a lungo termine Il numero di soggetti generalmente studiati è nelle centinaia/migliaia FASI della SPERIMENTAZIONE CLINICA FASE IV: è la stima accurata del rapporto sicurezza/efficacia in condizioni di usuale utilizzazione medica del trattamento. Il numero di soggetti generalmente studiati è nelle migliaia Valuta tollerabilità a lungo termine, effetti rari Di Orio, 1994. Pag132. Trials controllati e Randomizzati I trials controllati e randomizzati (RCTs), standard della sperimentazione terapeutica, rappresentano il disegno più affidabile da cui ricavare misure valide di efficacia per produrre raccomandazioni cliniche. • Gli esiti (outcomes) di qualunque malattia sono influenzati, oltre che dall’intervento terapeutico - da numerosi fattori: gravità e fluttuazioni della malattia, patologie concomitanti, fattori prognostici noti ed ignoti. • La randomizzazione assicura che tutti questi fattori vengano equamente distribuiti nei due gruppi di pazienti. • Se un RCT è condotto ed analizzato in maniera adeguata, l’eventuale differenza di esiti tra i due gruppi può essere ascritta all’effetto del trattamento in studio. • Gli studi che non effettuano un’assegnazione randomizzata dei pazienti al trattamento, sono contraddistinti da una percentuale maggiore di risultati falsamente positivi. PROTOCOLLO dello STUDIO Scopo della ricerca Eticità della sperimentazione Disegno dello studio Criteri di selezione (inclusione/esclusione o eleggibilità) Metodo di randomizzazione Fattori prognostici Variabile di risposta Numerosità del campione Metodi statistici analisi PROTOCOLLO dello STUDIO Scopo della ricerca Eticità della sperimentazione Disegno dello studio Criteri di selezione (inclusione/esclusione o eleggibilità) Metodo di randomizzazione Fattori prognostici Variabile di risposta Numerosità del campione Metodi statistici analisi Scopo della ricerca La ricerca deve essere impostata in modo da rispondere a quesiti che devono essere esplicitamente definiti e numericamente limitati. Ogni studio deve mirare a saggiare un obiettivo principale, di rilevante importanza clinica, su cui si impernia il calcolo della dimensione campionaria. Possono poi essere investigati altri obiettivi secondari, che devono essere in numero limitato e identificati all’inizio dello studio. PROTOCOLLO dello STUDIO Scopo della ricerca Eticità della sperimentazione Disegno dello studio Criteri di selezione (inclusione/esclusione o eleggibilità) Metodo di randomizzazione Fattori prognostici Variabile di risposta Numerosità del campione Metodi statistici analisi Eticità della sperimentazione La sperimentazione deve essere pianificata e condotta in accordo ai principi della dichiarazione di Helsinki e successivi emendamenti. Deve essere posta molta attenzione nella stesura del consenso informato. PROTOCOLLO dello STUDIO Scopo della ricerca Eticità della sperimentazione Disegno dello studio Criteri di selezione (inclusione/esclusione o eleggibilità) Metodo di randomizzazione Fattori prognostici Variabile di risposta Numerosità del campione Metodi statistici analisi Disegni di RCT Entro pazienti Tra i pazienti: - disegno randomizzato semplice - disegno randomizzato stratificato - disegno fattoriale Disegni di RCT Entro pazienti (Crossover) Ciascun paziente riceve in sequenza due o più trattamenti in esame. In questo caso è randomizzata la sequenza con cui i trattamenti sono somministrati a ciascun paziente. Attenzione all’effetto carryover! Disegni di RCT Tra i pazienti: - disegno randomizzato semplice - disegno randomizzato stratificato - disegno fattoriale DISEGNO RANDOMIZZATO SEMPLICE Di Orio, 1994. Pag124. DISEGNO RANDOMIZZATO STRATIFICATO Negli studi randomizzati stratificati disegnando lo studio si decide a priori di creare degli strati in cui i pazienti siano assegnati a caso in numero pari ai due trattamenti. Lo strato può essere rappresentato dal centro di reclutamento negli studi multicentrici oppure, in caso di malattie in cui non sia chiaro il ruolo prognostico di alcune variabili, lo strato può essere rappresentato dalle modalità delle variabili prognostiche stesse. DISEGNO FATTORIALE Negli studi fattoriali si decide di valutare contemporaneamente due o più obiettivi. La combinazione dei due obiettivi configura quattro gruppi di trattamento Es. il Gruppo Italiano Studio Sopravvivenza Infarto Miocardio ha disegnato un trial con un duplice obiettivo: a) valutare l’effetto di due trombolitici (streptochinasi e alteplase) in pz con infarto del miocardio entro sei ore dall’attacco b) valutare negli stessi pz l’eventuale effetto dell’eparina sottocute in aggiunta alla terapia tradizionale. In questo caso i gruppi di trattamento erano: 1) Terapia tradizionale + streptochinasi 2) Terapia tradizionale + streptochinasi + eparina 3) Terapia tradizionale + alteplase 4) Terapia tradizionale + alteplase + eparina PROTOCOLLO dello STUDIO Scopo della ricerca Eticità della sperimentazione Disegno dello studio Criteri di selezione (inclusione/esclusione o eleggibilità) Metodo di randomizzazione Fattori prognostici Variabile di risposta Numerosità del campione Metodi statistici analisi Criteri di selezione I criteri di inclusione identificano il gruppo di pazienti su cui sarà effettuato lo studio clinico e definiscono di conseguenza la popolazione obiettivo dello studio. I criteri di inclusione/esclusione dei pazienti possono essere intrinseci al paziente (es. sesso), alla patologia in studio (es. stadio del tumore) o legati a considerazione generali (es. patologie concomitanti). PROTOCOLLO dello STUDIO Scopo della ricerca Eticità della sperimentazione Disegno dello studio Criteri di selezione (inclusione/esclusione o eleggibilità) Metodo di randomizzazione Fattori prognostici Variabile di risposta Numerosità del campione Metodi statistici analisi Metodo di randomizzazione Semplice (numeri casuali) A blocchi Stratificata Minimizzazione Il metodo utilizzato deve essere sempre esplicitato PROTOCOLLO dello STUDIO Scopo della ricerca Eticità della sperimentazione Disegno dello studio Criteri di selezione (inclusione/esclusione o eleggibilità) Metodo di randomizzazione Fattori prognostici Variabile di risposta Numerosità del campione Metodi statistici analisi Fattori prognostici Occorre considerare tutti i fattori prognostici che si conoscono in letteratura e definire se saranno trattati in fase di pianificazione dello studio con stratificazione o successivamente in fase di analisi dei dati. PROTOCOLLO dello STUDIO Scopo della ricerca Eticità della sperimentazione Disegno dello studio Criteri di selezione (inclusione/esclusione o eleggibilità) Metodo di randomizzazione Fattori prognostici Variabile di risposta Numerosità del campione Metodi statistici analisi Variabile di risposta (endpoint) L’endpoint deve essere misurato nello stesso modo per i pazienti in trattamento ed i pazienti in controllo. Possibili endpoints: proporzione di complicanze, mortalità, proporzione di recidive, etc Gli endpoints possono essere clinici o surrogati End Points surrogati negli RCT Intervento Malattia End Point Surrogato Clinical Outcome Vero Fleming T, et al. Ann Intern Med 1996 Esempio di End Point surrogato Dieta ricca di flavonoidi Tumore della Vescica Mutazioni nella cellule esfoliate della vescica Presenza di cellule tumorali a livello vescicale PROTOCOLLO dello STUDIO Scopo della ricerca Eticità della sperimentazione Disegno dello studio Criteri di selezione (inclusione/esclusione o eleggibilità) Metodo di randomizzazione Fattori prognostici Variabile di risposta Numerosità del campione Metodi statistici analisi TIPI DI ERRORE NEI TEST STATISTICI • Errore di tipo I (alfa) • Probabilità di rifiutare l’ipotesi nulla quando questa è vera • Errore di tipo II (beta) • Probabilità di non rifiutare l’ipotesi nulla quando questa è falsa Che cos’è la potenza statistica? (1 – beta) La potenza è la probabilità di identificare un effetto di una certa entità predefinita del trattamento, quando essa esiste, ad un livello specifico di significatività statistica (per esampio p<0.05), e per una particolare numerosità campionaria. Numero di soggetti per gruppo che e’ necessario studiare in diverse condizioni per dimostrare una differenza tra proporzioni (alfa=0.05 a 2 code) Frequenza di eventi nel gruppo sperimentale: 5% 10% 20% Frequenza di eventi nel gruppo di controllo: Potenza statistica 25% 30% 40% 80% 49 36 22 95% 80 58 34 80% 100 62 32 95% 164 101 52 80% 1094 294 82 95% 1810 485 134 Numerosità campionaria come funzione della frequenza dell’esposizione e OR OR=1.2 3000 No. cases (=No.controls) 2750 2500 2250 OR=1.4 OR=1.5 OR=2.0 OR=3.0 2000 1750 1500 1250 1000 750 500 250 0 0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0 Prevalence of "at risk" genotype PROTOCOLLO dello STUDIO Scopo della ricerca Eticità della sperimentazione Disegno dello studio Criteri di selezione (inclusione/esclusione o eleggibilità) Metodo di randomizzazione Fattori prognostici Variabile di risposta Numerosità del campione Metodi statistici analisi INTENTION TO TREAT Misure di efficacia utilizzate dai trials Evento Rischio di sviluppare Presente Assente l’evento Trattati A B Y= A/(A+B) Controlli C D X= C/(C+D) Rischio dell’evento nei trattati Y= A/(A+B) Rischio dell’evento nei controlli X= C/(C+D) • Rischio Relativo RR= Y/X • Riduzione del Rischio Relativo RRR= (X-Y)/X x 100 • Odds Ratio OR= (A/B)/(C/D) • Riduzione del Rischio Assoluto RRA= X-Y • Numero Necessario da Trattare NNT= 1/(X-Y) Hypertension Optimal Treatment (HOT) Trial Evento Presente Assente Rischio di sviluppare l’evento Trattati 315 9084 Y= A/(A+B) = 0.033 Controlli 368 9023 X= C/(C+D) = 0.039 • Rischio Relativo RR= 0.85 (0.74 to 0.99) • Riduzione del Rischio Relativo RRR= 15% (1% to 26%) • Odds Ratio OR= 0.85 (0.71 to 0.99) • Riduzione del Rischio Assoluto RRA= 0.006 (0.0003 to 0.01) • Numero Necessario da Trattare NNT= 167 (90 to 3117)=1/RRA Hansson L, et al. Lancet 1998 RRA=0.039-0.033=0.006 NNT=1/0.006=167 Number Needed to Treat (NNT) -Incorpora il rischio di base del paziente e l’entità assoluta della riduzione del rischio - E' facile da calcolare e da utilizzare - Consente di esprimere nella stessa unità di misura (il paziente) sia i benefici che i rischi di un intervento preventivo - Fornisce una misura indicativa dei costi (diretti ed indiretti) di un intervento terapeutico o preventivo. - Consente di confrontare diversi interventi per pianificare le strategie di politica sanitaria Cartabellotta A, et al. Epidemiol & Pre 1997 Numero di pazienti da trattare (NNT) con misoprostolo per sei mesi per evitare una complicanza gastrointestinale severa Pazienti Tutti >65 aa >75 aa Tutti 264 220 300 Con pregressi eventi cardiovascolari 204 142 144 Con pregressa ulcera peptica 52 40 22 Con pregressa emorragia digestiva 40 32 14 Dati da: Silverstein FE, et al. Ann Intern Med 1995 Riduzione del rischio relativo ottenuta dal trattamento Rischio basale 50 40 30 25 20 15 10 0,9 2 3 4 4 6 7 11 0,6 3 4 6 7 8 11 17 0,3 7 8 11 13 17 22 33 0,2 10 13 17 20 25 33 50 0,1 20 25 33 40 50 67 100 0,05 40 50 67 80 100 133 200 0,01 200 250 333 400 500 667 1000 0,005 400 500 667 800 1000 1333 2000 0,001 2000 2500 3333 4000 5000 6667 10000 NNT Laupacis A et al. N Engl J Med 1988
Scaricare