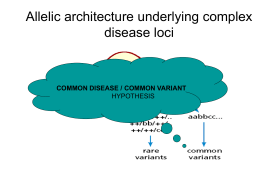
corso di Genomica 2010-2011 lezione 15-16 • laurea magistrale Biotecnologia Industriale Giovedì 2 dicembre 2010 aula 6 orario : Martedì ore 14.00 - 16.00 Giovedì ore 13.00 - 15.00 D. Frezza recupero lezione del 30 Nov. 2010 regalo per St.Nikolaos Lunedì 13 Dicembre seminario sui nuovi metodi di sequenziamento presso la facoltà di Medicina ore 9:00 aula anfiteatro (piano -1) seminario Lunedì 13 Dicembre sequencing new generation MARCO ISLAND, Fla. — Ion Torrent Systems unveiled an electronic sequencer last week that reads DNA on a semiconductor chip by measuring the release of hydrogen ions as nucleotides get incorporated by DNA polymerase. The instrument will cost less than $50,000 and generate "hundreds of millions of bases" and "millions" of highly accurate reads per run, each several hundred bases in length, according to Jonathan Rothberg, the company's co-founder and CEO. Each run will take about an hour and cost less than $500. Speaking in front of a packed audience at the end of the last session of the Advances in Genome Biology and Technology conference here, Rothberg said that the company, which is based in Guilford, Conn., and San Francisco and has been operating quietly since its foundation in 2007, plans to sell tens of thousands of the instruments to laboratories around the world. Although the system uses polymerase-based sequencing-by-synthesis chemistry — like most existing second-generation sequencers — it is the first to do away with lasers, cameras, or labels, relying entirely on electronic detection. "The machine is now a chip," Rothberg said. association studies Association studies using common allelic variants are cheaper and simpler than the complete resequencing of candidate genes, and have been proposed as a powerful means of identifying the common variants that underlie complex traits. In their simplest form, association studies compare the frequency of alleles or genotypes of a particular variant between disease cases and controls. Alternative approaches include using family-based controls to avoid the potential problem of population stratification. case control studies confronto di frequenze alleliche tra i soggetti patologici o con 1 fenotipo e le frequenze della popolazione di controllo problemi di stratificazione: come si devono scegliere le 2 popolazioni da controllare? I paesi oltreoceano hanno popolazioni miste (melting pot) e devono essere pesate le componenti della popolazione di controllo di riferimento e non tutti gli africani o gli europei sono uguali, cioè non basta prendere dei neri e dei bianchi come riferimento. Esiste una statistica apposta per questo: tests di caso-controlli una meta analisi fatta su dati di letteratura pubblicati: (andatevi a vedere cosa è una meta-analisi) Ethnic difference in patients with type 2 diabetes mellitus in inter-East Asian populations: a systematic review and meta-analysis focusing on gene polymorphism. Takeuchi M, Okamoto K, Takagi T, Ishii H. J Diabetes. 2009 Dec;1(4):255-62. METHODS: Data sources included MEDLINE and EMBASE between January 2001 and October 2008. We conducted a search for articles containing minor allele frequency (MAF) in the gene polymorphisms of peroxisome proliferator-activated receptor-γ (PPARG), inward-rectifying potassium channel Kir6.2 (KCNJ11), Calpain 10 (CAPN10), and transcription factor 7-like 2 (TCF7L2). The pooled odds ratio was calculated by using a fixed-effects model with the Mantel-Haenszel method after confirming statistical evidence of homogeneity across the ethnicities using the Breslow-Day test. candidate gene association limits Candidate-gene association studies have identified many of the genes that are known to contribute to susceptibility to common disease. Such studies are greatly facilitated by using indirect LINKAGEDISEQUILIBRIUM (LD)-based methods. However,candidate-gene studies rely on having predicted the identity of the correct gene or genes, usually on the basis of biological hypotheses or the location of the candidate within a previously determined region of linkage.Even if these hypotheses are broad (for example, involving the testing of all genes in the insulin-signalling pathway), they will, at best, identify only a fraction of genetic risk factors, even for diseases in which the pathophysiology is relatively well understood. When the fundamental physiological defects of a disease are unknown, the candidate-gene approach will clearly be inadequate to fully explain the genetic basis of the disease. genome wide association approach definizione: studio di associazione causale di varianti genetiche con una rassegna del genoma. Non ci sono preconcetti sulla regione genomica delle varianti. Il metodo sfrutta la forza dell’associazione senza avere una ipotesi sull’identità del gene causale. E’ un metodo non “bias” (sapete cosa vuol dire?) cioè privo di una preferenza di scelta, anche in presenza di evidenze convincenti contrarie sulla funzione e localizzazione dei geni causali. Deve essere un metodo capace di trovare appunto i geni che potrebbero sfuggire ad una indagine del tipo gene-candidato in cui si suppone l’associazione di un metabolismo ai suoi geni correlati come predisponenti. Qui è l’opposto: ricerca dei geni non correlabili sulla base delle evidenze note. base statistica per WGS Estimating haplotype frequencies by combining data from large DNA pools with database information. We assume that allele frequency data have been extracted from several large DNA pools, each containing genetic material of up to hundreds of sampled individuals. Our goal is to estimate the haplotype frequencies among the sampled individuals by combining the pooled allele frequency data with prior knowledge about the set of possible haplotypes. Such prior information can be obtained, for example, from a database such as HapMap. We present a Bayesian haplotyping method for pooled DNA based on a continuous approximation of the multinomial distribution. The proposed method is applicable when the sizes of the DNA pools and/or the number of considered loci exceed the limits of several earlier methods. In the example analyses, the proposed model clearly outperforms a deterministic greedy algorithm on real data from the HapMap database. With a small number of loci, the performance of the proposed method is similar to that of an EMalgorithm, which uses a multinormal approximation for the pooled allele frequencies, but which does not utilize prior information about the haplotypes. The method has been implemented using Matlab and the code is available upon request from the authors. Gasbarra D, Kulathinal S, Pirinen M, Sillanpää MJ. University of Helsinki, Helsinki. IEEE/ACM Trans Comput Biol Bioinform. 2011 Jan-Mar;8(1):36-44. perchè Genome Wide ass. studies “approaches to mapping the genes that underlie common disease and quantitative traits fall into two categories: CANDIDATE-GENE studies, which use either association or resequencing approaches, and genome-wide studies, which include both LINKAGE MAPPING and genome-wide association studies. The approaches and their advantages and disadvantages are summarized in TABLE 1. In this review,we discuss these approaches and present arguments as to why genome wide association studies might be advantageous for identifying the genetic variants associated with common disease. One fundamentally different approach, ADMIXTURE MAPPING**, is not discussed here but has been described elsewhere7–10.” ** studi su campioni di popolazioni mescolate es: America (USA, Brasile ecc.) il problema è il controllo di riferimento Hap Map project Differences in individual bases are by far the most common type of genetic variation. These genetic differences are known as single nucleotide polymorphisms, or SNPs (pronounced "snips"). By identifying most of the approximately 10 million SNPs estimated to occur commonly in the human genome, the International HapMap Project is identifying the basis for a large fraction of the genetic diversity in the human species. However, testing all of the 10 million common SNPs in a person's chromosomes would be extremely expensive. The development of the HapMap will enable geneticists to take advantage of how SNPs and other genetic variants are organized on chromosomes. Genetic variants that are near each other tend to be inherited together. For example, all of the people who have an A rather than a G at a particular location in a chromosome can have identical genetic variants at other SNPs in the chromosomal region surrounding the A. These regions of linked variants are known as haplotypes (Figure 2). figura 2 SNPs la combinazione dei singoli polimorfismi forma un aplotipo QuickTime™ e un decompressore TIFF (Non compresso) sono necessari per visualizzare quest'immagine. didascalia fig.2 aplotipi Figure 2: The construction of the HapMap occurs in three steps. (a) Single nucleotide polymorphisms(SNPs) are identified in DNA samples from multiple indivduals. (b)Adjacent SNPs that are inherited together are compiled into "haplotypes." (c)"Tag" SNPs within haplotypes are identified that uniquely identify those haplotypes. By genotyping the three tag SNPs shown in this figure, researchers can identify which of the four haplotypes shown here are present in each individual. finalità del prog. HapMap In many parts of our chromosomes, just a handful (manciata) of haplotypes are found in humans. [See The Origins of Haplotypes: http://snp.cshl.org/originhaplotype.html] In a given population, 55 percent of people may have one version of a haplotype, 30 percent may have another, 8 percent may have a third, and the rest may have a variety of less common haplotypes. The International HapMap Project is identifying these common haplotypes in four populations from different parts of the world. It also is identifying "tag" SNPs that uniquely identify these haplotypes. By testing an individual's tag SNPs (a process known as genotyping), researchers will be able to identify the collection of haplotypes in a person's DNA. The number of tag SNPs that contain most of the information about the patterns of genetic variation is estimated to be about 300,000 to 600,000, which is far fewer than the 10 million common SNPs. formazione degli aplotipi Over the course of many generations, segments of the ancestral chromosomes in an interbreeding population are shuffled (mescolati) through repeated recombination events. Some of the segments of the ancestral chromosomes occur as regions of DNA sequences that are shared by multiple individuals (Figure 1). These segments are regions of chromosomes that have not been broken up by recombination, and they are separated by places where recombination has occurred. These segments are the haplotypes that enable geneticists to search for genes involved in diseases and other medically important traits. utilità degli aplotipi The fossil record and genetic evidence indicate that all humans today are descended from anatomically modern ancestors who lived in Africa about 150,000 years ago. Because we are a relatively young species, most of the variation in any current human population comes from the variation present in the ancestral human population. Also, as humans migrated out of Africa, they carried with them part but not all of the genetic variation that existed in the ancestral population. As a result, the haplotypes seen outside Africa tend to be subsets of the haplotypes inside Africa. In addition, haplotypes in non-African populations tend to be longer than in African populations, because populations in Africa have been larger through much of our history and recombination has had more time there to break up haplotypes. figura meiosi X over Figure 1: This diagram shows two ancestral chromosomes being scrambled through recombination over many generations to yield different descendant chromosomes. If a genetic variant marked by the A on the ancestral chromosome increases the risk of a particular disease, the two individuals in the current generation who inherit that part of the ancestral chromosome will be at increased risk. Adjacent to the variant marked by the A are many SNPs that can be used to identify the location of the variant. QuickTime™ e un de com press ore TIFF (No n compre sso) so no n ece ssari per vi sual izza re qu est'imm agin e. dispersione degli aplotipi As modern humans spread throughout the world, the frequency of haplotypes came to vary from region to region through random chance, natural selection, and other genetic mechanisms. As a result, a given haplotype can occur at different frequencies in different populations, especially when those populations are widely separated and unlikely to exchange much DNA through mating. Also, new changes in DNA sequences, known as mutations, have created new haplotypes, and most of the recently arising haplotypes have not had enough time to spread widely beyond the population and geographic region in which they originated. applicazioni di HapMap Once the information on tag SNPs from the HapMap is available, researchers will be able to use them to locate genes involved in medically important traits. Consider the researcher trying to find genetic variants associated with high blood pressure. Instead of determining the identity of all SNPs in a person's DNA, the researcher would genotype a much smaller number of tag SNPs to determine the collection of haplotypes present in each subject. The researcher could focus on specific candidate genes that may be associated with a disease, or even look across the entire genome to find chromosomal regions that may be associated with a disease. If people with high blood pressure tend to share a particular haplotype, variants contributing to the disease might be somewhere within or near that haplotype.
Scaricare