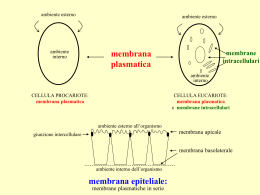
Il destino dei farmaci nell’organismo Concentrazione del farmaco L’azione dei farmaci dipende dalla concentrazione raggiunta al sito d’azione. Tale concentrazione costituisce la variabile quantitativa principalmente considerata dalla farmacologia per spiegare l’azione dei farmaci. Farmacologia • Farmacocinetica: studio degli eventi che il farmaco subisce all’interno dell’organismo che regolano la concentrazione del farmaco • Farmacodinamica: studio delle interazioni tra I farmaci ed i loro recettori molecolari Quantita’ di farmaco somministrata Dose Volume di distribuzione Concentrazione del farmaco al sito attivo Farmacocinetica Farmacodinamica Processi che influenzano il destino dei farmaci nell’organismo • • • • Assorbimento al sito di somministrazione Distribuzione nell’organismo Metabolismo (biotrasformazione) Eliminazione ADME Attraversamento delle membrane biologiche da parte dei farmaci I farmaci per esplicare la loro azione farmacologica devono in genere attraversare diverse membrane biologiche. La capacità dei farmaci di attraversare le membrane influenza tutti i processi farmacocinetici (assorbimento e distribuzione prima di tutto, ma anche metabolismo ed eliminazione/escrezione) Attraversamento barriere cellulari Un farmaco può essere efficace solo se e’ in grado di superare le barriere presenti nell’organismo e quindi di distribuirsi. Il passaggio di un farmaco attraverso le membrane cellulari dipende dalle seguenti caratteristiche: - dimensione/struttura della molecola; - liposolubilita’; - grado di ionizzazione/dissociazione. Dimensione molecolare Coefficiente di diffusione è inversamente proporzionale alla radice quadrata del peso molecolare. La dimensione molecolare influenza in minima parte le caratteristiche farmacocinetiche di «small molecular entities» (200 – 1000 Da), mentre ha un ruolo importante per i farmaci biologici costituiti da macromolecole. Modalità di passaggio dei farmaci attraverso le membrane cellulari: piccole molecole Membrana cellulare Spessore compreso fra 7 e 12 nm Formata da un doppio strato di fosfolipidi, attraversato parzialmente o completamente da proteine. Modalità di attraversamento delle barriere cellulari da parte dei farmaci Modalità di passaggio dei farmaci attraverso le membrane cellulari: macromolecole Liposolubilità Si può misurare utilizzando il coefficiente di ripartizione tra una fase idrofoba ed una idrofila (per esempio, coefficiente di ripartizione ottanolo acqua). Importanza della liposolubilità nel passaggio delle membrane Il numero di molecole che attraversano la membrana per unità di superficie e nell’unità di tempo è dato dal coefficiente di permeabilità e dalla differenza di concentrazione fra i due lati della membrana Attraversamento dei farmaci delle membrane biologiche (formula generale) dM/dt = [D x A x (K1 C1 – K2 C2)] / h dM/dt = flusso attraverso la membrana D = coefficiente di diffusione (dipende dalle dimensioni della molecola) A = superficie di assorbimento K1 = coefficiente di ripartizione fra membrana e fluido donatore C1 = concentrazione in fluido donatore K2 = coefficiente di ripartizione fra membrana e fluido accettare C2 = concentrazione in fluido accettore h = spessore membrana Attraversamento dei farmaci delle membrane biologiche (formula generale) dM/dt = [D x A x K x (C1 – C2)] / h dM/dt = flusso attraverso la membrana D = coefficiente di diffusione (dipende dalle dimensioni della molecola) A = superficie di assorbimento K = coefficiente di ripartizione fra membrana e acqua C1 = concentrazione in fluido donatore C2 = concentrazione in fluido accettore h = spessore membrana Lipofilicità e coefficiente di ripartizione A parità di gradiente di concentrazione, una sostanza lipofila (coefficiente di ripartizione K elevato) diffonderà più velocemente rispetto una sostanza con coefficiente di ripartizione K ridotto. Dissociazione dei farmaci (acido/base) e loro distribuzione Molti farmaci sono dal punto di vista chimico acidi o basi deboli e quindi possono trovarsi nei fluidi biologici nella forma ionizzata o non ionizzata. La forma ionizzata presenta una liposolubilità molto bassa e non è in genere in grado di attraversare le membrane (ad esclusione di casi in cui è presente un meccanismo di trasporto specifico). Dissociazione dei farmaci (acido/base) e loro distribuzione [𝑓𝑜𝑟𝑚𝑎 𝑝𝑟𝑜𝑡𝑜𝑛𝑎𝑡𝑎] 𝐿𝑜𝑔 = 𝑝𝐾𝑎 − 𝑝𝐻 [𝑓𝑜𝑟𝑚𝑎 𝑛𝑜𝑛 𝑝𝑟𝑜𝑡𝑜𝑛𝑎𝑡𝑎] pKa rappresenta il pH a cui metà del farmaco (acido debole o base debole) si trova in forma ionizzata Forma protonata = HA o BH+ Forma non protonata = A- o BH Dissociazione dei farmaci (acido/base) e attraversamento membrane Influence of pH on the distribution of a weak acid between plasma and gastric juice separated by a lipid barrier. A. The dissociation of a weak acid, pKa = 4.4. B. Dissociation of the weak acid in plasma (pH 7.4) and gastric acid (pH 1.4). The uncharged from, HA, equibrates across the membrane. Blue numbers in brackets show relative concentrations of HA and A−. Dissociazione dei farmaci (acido/base) e loro distribuzione Dissociazione dei farmaci (acido/base) e loro distribuzione I farmaci acidi (pKa < 7) tendono a concentrarsi nei compartimenti basici, mentre i farmaci basici (pKa > 7) tendono a concentrarsi nei compartimenti acidi. Queste proprieta’ possono essere sfruttare per facilitare l’eliminazione dei farmaci dall’organismo con l’alcalinizzazione delle urine (per farmaci acidi, e.g., salicilati). Dissociazione dei farmaci (acido/base) e loro distribuzione Acido acetil salicilico (FANS) petidina/meperidina (analgesico oppioide)
Scaricare