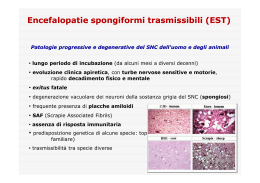
Le malattie da PRIONI Io…paura della mucca pazza?? Perché dovrei???? sono un gallo!!!! I prioni Il termine di prioni è stato coniato per indicare agenti patogeni di natura proteica provvisti di proprietà infettanti, in grado di provocare una serie di encefalopatie degenerative dell’uomo e di alcuni animali. Le malattie da prioni: malattie “genetiche” o malattie “da infezione”? • I prioni sono isoforme patologiche di proteine normali (presenti soprattutto nei neuroni) che hanno la loro origine nella presenza di mutazioni nel gene codificatore e che si accumulano nelle cellule neuronali, danneggiandole irreversibilmente. Le proteine prioniche, pur essendo il risultato di una mutazione genica, sono capaci di moltiplicarsi nell’ospite e di indurre una patologia……….. • L’inoculazione o l’ingestione di sospensioni contenenti prioni è in grado di indurre la comparsa della patologia in soggetti normali, i quali a loro volta diventano sorgente di possibile infezione. Le encefalopatie spongiformi I prioni sono gli agenti responsabili di una serie di encefalopatie spongiformi caratterizzate, dal punto di vista istologico, dalla presenza di lesioni degenerative, che conferiscono al tessuto nervoso un aspetto spongioso (spongiforme: comparsa di vacuoli a livello dei dendriti neuronali e dei neuroni) • • • • Si manifestano con: atrofia perdita di cellule neuronali proliferazione delle cellule gliali deposizione di fibrille di natura glicoproteica (placche) Sistema nervoso normale Patologico (vacuolo all’interno del nervo facciale) TSE Illustrazione schematica delle più frequenti localizzazioni delle lesioni da malattie prioniche. prioniche. Nella BSE è colpito il midollo allungato, nella FFI la regione talamica, nel CJD la corteccia cerebrale, mentre nel KURU e nella GSS viene maggiormente interessato il cervelletto. TSE TSE umane non si trasmettono per via verticale. Alcune (GSS e 10% di CJD ) hanno meccanismo di tipo ereditario Gruppo malattie eterogeneo Trasmissione verticale probabile per Scrapie TSE Lungo periodo di incubazione Sintomatologia di tipo neurologico Decorso clinico progressivo e costantemente fatale Lesioni di tipo degenerativo al SNC No lesioni di tipo infiammatorio o risposte immunitarie Costante presenza di una proteina specifica (PrPres (PrPres o PrPsc) PrPsc) responsabile delle lesioni degenerative TSE PRIONE: fattori patogeneticamente importanti Mancata risposta immunitaria o infiammatoria Elevata resistenza alla degradazione Estrema resistenza a numerosi agenti fisici o chimici in grado di inattivare altri microorganismi TRATTAMENTI INEFFICACI PER INATTIVARE GLI AGENTI INFETTANTI TSE AGENTI FISICI AGENTI CHIMICI Calore umido (100°C per 1 ora) Etanolo Congelamento Formaldeide Radiazioni ultraviolette Acqua ossigenata Radiazioni ionizzanti Iodofori pH 2,1 - 10,5 Permanganato Disinfettanti fenolici Procedure in grado di inattivare agenti TSE Procedure ad alta efficienza Temperatura o concentrazione Tempo ipoclorito di sodio 2% di Cloro attivo 1 ora Autoclave in NaOH (idrossido di Na) 2M 121°C 30 minuti Acido formico (tessuto cerebrale fissato in formalina) 98% 1 ora Transmissible Spongiform Encephalopathy • Le TSE sono attualissime, dopo l’epidemia BSE nel Regno Unito (anni 1985 – 1995 ) • La scrapie è la TSE meglio conosciuta (TSE prototipo) Le encefalopatie spongiformi sono trasmissibili Scrapie: è stata la prima encefalopatia spongiforme di cui è stata dimostrata l’infettività, manifestatasi nelle greggi di pecore (1936). Si manifesta clinicamente con mancanza di coordinamento muscolare (atassia), tremori, deperimento organico (cachessia) e intenso prurito. La trasmissione orizzontale fu dimostrata per la prima volta nel 1936 da Cullié e Chelle. Le encefalopatie spongiformi sono trasmissibili Scrapie: L’infezione avviene attraverso la via orale. La trasmissione può avvenire sia per via orizzontale (principalmente durante i parti) sia, probabilmente, per via materna (???). La malattia può essere diffusa da animali infetti ma ancora asintomatici. Inoltre, sembra sussistere una relazione dose-risposta: tanto più lunga (e precoce) è l’esposizione degli animali sani ad animali infetti tanto maggiore è l’incidenza nell’allevamento, con un accorciamento dei tempi di incubazione Le encefalopatie spongiformi sono trasmissibili Kuru: atassia progressiva dimostrata nella Nuova Guinea (1950). C. Gajdusek dimostrò che le lesioni encefaliche presenti nei soggetti affetti da Kuru erano identiche a quelle presenti negli animali infetti da scrapie. Inoltre, l’inoculazione intracerebrale di materiale cerebrale proveniente da soggetti deceduti per kuru provocava nello scimpanzé manifestazioni cliniche e anatomo-patologiche identiche. 12-18 mesi Trasmissione orrizontale Le encefalopatie spongiformi riguardanti la patologia animale •Scrapie (pecore) •Cachessia cronica (alce, cervo) •Encefalopatia spongiforme dei felini •Encefalopatia trasmissibile del visone •Encefalopatia spongiforme del bovino (BSE) sembrano tutte imputabili ad infezioni esogene contratte per via alimentare, in seguito alla introduzione nella catena alimentare animale, di materiali provenienti da animali portatori di patologie di questo tipo Le encefalopatie spongiformi riguardanti la patologia umana • SPORADICHE : CJD (malattia di Creutzfeldt-Jacob) 1 caso ogni 2.000.000 persone/anno. Alterazione spontanea PrP? • FAMILIARI: Malattia di Gerstmann-Straussler-Scheinker; Insonnia Familiare Fatale. Alterazione trasmissibile PrP? • INFETTIVE: KURU; nv CJD (variante della malattia di Creutzfeldt-Jacob) Insonnia Familiare Fatale (FFI) • • Età media: 40 anni. A differenza di altre encefalopatie spongiformi, i cambiamenti della sostanza grigia sono confinate ai nuclei talamici e provocano disturbi del ritmo sonno-veglia. • Negli stadi precoci della malattia, il paziente può avere difficoltà nell’addormentarsi e difficoltà motorie intermittenti. Questo stadio può durare mesi, ma alla fine progredisce nell’insonnia, in iperattività simpatica e nella demenza. • Il corso della malattia dura in media 13 mesi I Ricercatori del Service de Neurologie dell’Hopital Saint-Joseph a Parigi hanno segnalato il 5° caso di insonnia familiare fatale in Francia, caratterizzato da una mutazione al codone 178 del gene della proteina prionica e da una condizione di eterozigosi Met/Val al codone 129. Malattia di Gerstmann-Straussler-Scheinker (GSS) • • • • Simile alla malattia di Creutzfeldt-Jakob, è trasmissibile ad animali da esperimento. La malattia è riscontrata ovunque; ad ogni modo, l’incidenza è di circa 100 volte inferiore rispetto alla malattia di Creutzfeldt-Jakob. Età di esordio più precoce (40 anziché 60 anni) e una più lunga durata media di malattia (5 anni anziché 9 mesi). I pazienti presentano una degenerazione spinocerebellare o una degenerazione olivo ponto cerebellare, con un’atassia cerebellare che si verifica precocemente. La malattia progredisce verso un’atassia degli arti, disartria, demenza, parkinsonismo, sordità, cecità e paralisi dello sguardo. Infine si ha una compromissione del tratto corticospinale. Malattia di Creutzefeldt-Jacob (CJ) CJ sporadica, familiare, iatrogena e nv diffusa in tutto il mondo con incidenza di: 1 caso/anno/106 individui. • • • Declino delle capacità motorie e cognitive, rapida (dal momento dei sintomi) evoluzione letale della patologia. Nella maggior parte dei casi si presenta in casi isolati (CJ sporadica), mentre occasionalmente (10%) nella forma familiare (CJ familiare-1924) con trasmissione ereditaria, autosomica, dominante. 1970: dimostrazione della trasmissibilità interumana, in conseguenza di interventi sanitari: verifica di alcuni casi di CJ iatrogena in pazienti trattati con ormoni della crescita, in soggetti sottoposti ad allotrapianti di cornea o in pazienti trattati con elettrodi stereotassici. Encefalopatia spongiforme del bovino (BSE) • • • • • 1986: Gran Bretagna. Origine: dallo scrapie (salto di specie??) Utilizzo di farine animali Paventato rischio trasmissione bovino-uomo. 1995-96 ad oggi: una nuova forma di CJ: Creutzefeldt-Jacob new-variant Creutzefeldt-Jacob New Variant • • • Sintomi psichiatrici: decorso relativamente protratto (fino a 38 mesi dall’inizio della sintomatologia alla morte del paziente). Insorge spontaneamente (in assenza di possibili cause di trasmissione iatrogena) in soggetti relativamente giovani (età media 27 anni) rispetto alla CJ sporadica (età media 65 anni). Le evidenze epidemiologiche (patologia riscontrata solo nelle popolazioni interessate dalla epidemia di BSE) e alcune caratteristiche del prione fanno considerare la CJ-nv come infezioni esogena contratta per via alimentare. I prioni…..eziologia L’ agente eziologico si dimostra resistente a trattamenti 1) in grado di inattivare i virus convenzionali 2) trattamenti in grado di inattivare gli acidi nucleici sensibile a trattamenti 1) (urea, fenoli) in grado di inattivare le proteine. Eziologia • La trasmissibilità sperimentale della patologia coincide con una proteina, parzialmente glicosilata formata da 254 aminoacidi e dal p.m. di 33-35kd, denominata PrP (proteina prionica) Questa proteina è formata prevalentemente da zone strutturali ad α-elica, lega atomi di rame nell’octarepeat (N(N-term) e svolge numerose funzioni nei fenomeni di plasticità neuronale. a elica b elica PrPSc proteina prionica associata allo scrapie e PrPc proteina prionica cellulare • Tutte le cellule dei mammiferi contengono un gene Prnp (cromosoma 20) che codifica una proteina molto simile chiamata PrPc Prnp PrPc • La PrPc codificata dal gene Prnp è costituita da 254 aminoacidi, è glicosilata a livello di due asparagine in posizione 181 e 197. La PrPc si ancora (mediante un gruppo glicosil-fosfadil-inositolo) alla membrana delle cellule (neuroni, polmoni, cuore, rene, pancreas, testicoli, leucociti e piastrine). • PrPSc proteina prionica patologica e PrPc proteina prionica cellulare La PrPSc e la PrPc differenze PrPc PrPSc dimensioni 33-35kd 27-30kd c.molecolare a helix b helix Resistenza alla digestione con enzimi proteolitici (proteasi) La “PrPc”(Prion Protein Cellular) è una proteina solubile le cui funzioni non sono ancora ben note ma importante nella trasmissione dei segnali nervosi. La “PrPsc”(Prion Protein Scrapie), è una proteina misfolded, in cui si ha la conversione delle α-eliche in foglietti β, cambiamento che provoca un ripiegamento diverso della proteina che la rende insolubile e resistente alle proteasi. Nel cervello queste proteine misfolded causano il cambiamento della proteina prionica nativa, PrPc, nella forma PrPsc. La proteina prionica PrPsc porta alla formazione di aggregati proteici, chiamate fibrille amiloidali che possono formare delle placche che sembrano apparire al microscopio come dei buchi al cervello. Questo causa deterioramento delle capacità fisiche e mentali, e alla fine la morte. Trasmissione sperimentale di prioni 1° inoculazione da una specie ad un altra p.i. lungo Passaggio successivo in animali della stesse specie Comparsa di sintomi morbosi p.i. più breve Se tentiamo di nuovo di inoculare nella specie di origine abbiamo lo stesso andamento, periodo lungo di incubazione prima della comparsa dei sintomi morbosi Barriera di specie…..e…. Barriera di specie…..e…. la sequenza della Prpsc è sempre quella della Prpc codificata dalle cellule dell’animale ospite e non la Prpsc utilizzata nell’inoculo iniziale e questo indica che.. La trasmissibilità dei prioni è un fenomeno che richiede la cooperazione di Prpc codificata dall’ospite con Prpsc esogena Trasmissione efficiente La barriera di specie Trasmissione inefficiente criceto 1: topo nel cui genoma è inserito il gene Prnp di criceto criceto Topo t1 Topo Topo criceto La barriera di specie è superata se nell’animale sono presenti geni eterologhi in grado di codificare la PrPc di origine Modello sperimentale di trasmissione dei prioni Encefalopatie umane o animali trasmissibili: • PrPc viene usualmente sintetizzata e degradata durante il normale metabolismo. Possono avvenire in modo assolutamente casuale delle modificazioni che portano la PrPc a forme parzialmente srotolate (PrP*) che rappresenterebbero una fase intermedia verso la transizione b sheet Prione esogeno PrPc degradazione PrP* PrPsc Modello sperimentale di trasmissione dei prioni • I prioni esogeni P legandosi ad una molecola PrP* * dell’ospite ne catalizzano la definiva transizione verso la forma patologica, con la produzione finale di due molecole di PrPsc. P * * P P P P accumulo P P P Dopo un certo periodo (incubazione) sufficiente all’accumulo delle quantità di proteina prionica patologica compaiono i sintomi Encefalopatie spongiformi a carattere familiare • • Si ha una trasmissione ereditaria autosomica e dominante. Nei soggetti portatori di una mutazione nel gene che codifica la PrP, la PrPc presenta delle mutazioni che diminuiscono la probabilità di assumere un normale aspetto. Le proteine prioniche neoformate intervengono nel passaggio PrP* ---PrPsc PrPc PrP* PrPsc P P PP P Encefalopatie spongiformi: forme sporadiche • Non sono mai state riscontrate mutazioni del gene. • La patologia potrebbe essere il risultato di mutazioni somatiche (che occorrono solo nelle cellule neuronali coinvolte dalla patologia) o di eventi casuali non noti. Encefalopatie spongiformi: CJ nuova variante Sono dovuti all’infezione da prioni esogeni, introdotti nell’organismo per via alimentare. Le evidenze: 1) correlazione spazio-temporale con l’ epidemia BSE negli animali 2) il prione della BSE è facilmente trasmissibile a topi transgenici per la proteina PrPc bovina 3) il prione della CJ nuova variante è trasmissibile ai topi nei quali provoca una patologia identica a quella indotta dal prione di origine bovina, risultando estremamente affine. Predisposizione genetica alle malattie da prioni • Una aumentata predisposizione genetica è individuabile nel polimorfismo presente a livello del codone (posizione 129) nel gene Prnp dove può essere normalmente presente sia il codone per l’aa valina (VAL), sia il codone per l’aa metionina (MET). Popolazione generale 37% Met/Met 51% Met/Val 12% Val/Val CJ sporadica la frequenza di omozigosi Val/Val Met/Met è del 90% Fattore di rischio Predisposizione genetica alle malattie da prioni • • • L’omozigosi non è un elemento predittivo (Giappone: elevata frequenza di Met/Met) ma un fattore predisponente in grado di agire in concomitanza con l’esposizione con altri fattori (?) essenziali per la comparsa della patologia. Una omozigosi Met/Met è comunque associata ad una forma di demenza rapida e progressiva Una omozigosi Val/Val è associata ad un decorso prolungato con prevalenti sintomi iniziali di atassia. CJ iatrogena • • Una omozigosi Met/Met o Val/Val è stata dimostrata anche in casi di CJ iatrogena associata alla somministrazione di ormone della crescita di origine estrattiva umana Una omozigosi Met/Met è stata dimostrata in tutti i casi di CJ nuova variante Quindi Il polimorfismo a livello del codone 129 svolge un ruolo genetico predisponente anche nei confronti della patologia da prioni esogeni, essendo il grado di influenzare l’interazione tra proteina prionica patologica e proteina normale Rischio di trasmissione orizzontale delle encefalopatie Una significativa trasmissibilità delle malattie da prioni è stata accertata nello scrapie delle pecore e delle capre (assunzione di annessi embrionali, per via alimentare, tra animali dello stesso gregge). Gli altri casi sono dovuti ad interventi artificiali: • cannibalismo nel Kuru • interventi sanitari nella CJ iatrogena • farine proteiche animali nella BSE CJD: infettività mediante sangue? E’ stato dimostrato che il sangue di animali infettati sperimentalmente può contenere livelli –bassi-dell’agente infettante identificato come causa di CJD, anche se al momento non si hanno prove certe della trasmissione tra umani. Ma…”l’assenza di evidenza non è evidenza di assenza” L’assenza di dati relativi alla trasmissione via sangue potrebbe essere dovuta a: 1. Assenza di infettività – plasma - prima dell’inizio della fase sintomatica e bassi livelli di infettività durante la fase sintomatica. 2. La necessità di una “carica infettante” di almeno 5 volte superiore a quella necessaria per la trasmissione via intracerebrale CJD: infettività mediante sangue? • • Al momento, non ci sono casi accertati casi CJ iatrogena (via sangue), anche se sono note donazioni effettuate da persone con CJD. Il rischio potenziale potrebbe essere variabile da 1/500.000 a 1/100.000.000. In realtà i trattamenti eseguiti durante la processazione del sangue sottolineano un rischio uguale a 0. Precauzioni • • • Impiego di prodotti (farmaci cosmetici) contenenti materiali di origine bovina) Impiego sanitario di materiali di origine umana (sangue, organi, tessuti) Trattamento ed eliminazione di materiali di origine umana in cui si sospetti il rischio (una notevole garanzia è rappresentata dal trattamento con NaOH 1N per 60 min e successivo trattamento in autoclave a 121°C per 30 min) Diagnosi etiologica • Il sospetto deriva da un progressivo declino delle funzioni cognitive e/o motorie del paziente . Non sono presenti fenomeni infiammatori, non si ha produzione di anticorpi LA DIAGNOSI E’ CLINICA • Diagnosi eziologica: Materiale bioptico cerebrale ricerca di PrPsc mediante antisieri specifici. Diagnosi etiologica I test rapidi attualmente accettati dalla Comunità Europea sono: Nome commerciale Metodica Ditta produttrice Prionics-Check Western Immunoblotting Prionics AG Enfer TSE ELISA in chemiluminescenza Abbott TeSeE ELISA sandwich Bio-rad Prionics-Check LIA ELISA in chemiluminescenza Prionics AG In Pro CDI-5 test Immunoassay conformazionale In Pro Biotechnology
Scarica