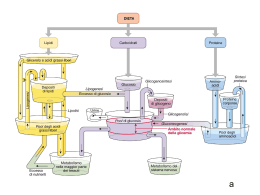
DISFUNZIONI MITOCONDRIALI E DIABETE DI TIPO 2 Lowel e Shulman (2005) Science Vol. 307 (allegato in formato PDF) Il diabete di tipo 2 è la malattia metabolica più diffusa nel mondo. Il diabete di tipo 2 sta rapidamente diventando una pandemia e si prevede che nel 2025 saranno oltre 300 milioni gli individui affetti nel mondo con un marcato aumento dei nuovi casi in India e in Asia. Malgrado la causa principale di questa malattia sia ancora sconosciuta, è chiaro che l’insulino-resistenza gioca un ruolo essenziale nella patogenesi e che un difetto nella secrezione di insulina da parte delle cellule β è strumentale nella progressione verso l’iperglicemia. In questa review viene formulata l’ipotesi che insulino-resistenza e iperglicemia siano entrambe attribuibili a difetti mitocondriali. RUOLO DEL METABOLISMO DEGLI ACIDI GRASSI NELL’INSULINORESISTENZA Molte evidenze indicano che l’insulino-resistenza sia il tratto principale del diabete di tipo 2. Infatti, tutti i pazienti con diabete di tipo 2 sono insulino-resistenti e studi prospettici hanno dimostrato che questa insulino-resistenza si sviluppa 1-2 decadi prima della malattia. Inoltre, l’insulino-resistenza nei figli di genitori affetti da diabete di tipo 2 è il miglior fattore predittivo per lo sviluppo successivo della malattia. Infine, fattori che riducono l’insulino-resistenza prevengono lo sviluppo del diabete. Il muscolo scheletrico ed il fegato sono i due organi chiave per il mantenimento della normale omeostasi del glucosio e la loro transizione ad uno stato di insulinoresistenza spiega la maggior parte delle alterazioni del metabolismo glucidico osservato nei pazienti con diabete di tipo 2. Prima di considerare se le disfunzioni mitocondriali contribuiscano o meno allo sviluppo dell’insulino-resistenza in questi organi, è opportuno capire i meccanismi cellulari responsabili dell’insulino- resistenza. Come discusso da Lazar et al. ci sono evidenze sempre più numerose che le citochine circolanti secrete dal tessuto adiposo possano modulare la risposta all’insulina del muscolo e del fegato. Comunque i metaboliti degli acidi grassi quali gli acilCoA, il diacilglicerolo o le ceramidi si pensa possano avere un ruolo critico. Circa 40 anni fa, Randle et al. dimostrarono che gli acidi grassi causavano insulinoresistenza in una preparazione in vitro di muscolo di ratto, ed essi ipotizzarono che questo avvenisse per un meccanismo di competizione di substrato. In accordo con questo modello, l’aumentata ossidazione degli acidi grassi nel muscolo produce un aumento dei livelli intracellulari di acetilCoA e di citrato, che possono di conseguenza inibire rispettivamente due enzimi coinvolti nell’utilizzo del glucosio: la piruvato deidrogenasi e la fosfofruttochinasi. L’inibizione del pathway glicolitico in questi step potrebbe incrementare il glucosio intracellulare e la concentrazione di G6P, che in ultimo determina una riduzione dell’assunzione di glucosio in risposta all’insulina. Tuttavia, studi più recenti usando il 13 C e il 31 P nella spettroscopia di risonanza magnetica (MRS) hanno dimostrato che questo meccanismo dell’insulinoresistenza indotta dagli acidi grassi nel muscolo scheletrico umano non regge, piuttosto gli acidi grassi sembrano causare insulino-resistenza attraverso la diretta inibizione dell’attività di trasporto del glucosio stimolata dall’insulina. Questa inibizione è probabilmente dovuta all’accumulo intracellulare di acilCoA e diacilglicerolo, i quali poi attivano pathway di trasduzione del segnale critici che alla fine portano alla soppressione del segnale dell’insulina (Figura 1). Quindi si potrebbe pensare che qualsiasi perturbazione metabolica che promuova l’accumulo di acidi grassi nel fegato e/o nel muscolo e/o ogni difetto nella capacità di questi organi di metabolizzare gli acidi grassi potrebbe indurre insulino-resistenza. Inoltre, difetti nel metabolismo degli adipociti, che si verificano in condizioni quali la severa lipodistrofia (mancanza di tessuto adiposo), possono determinare la prima condizione, mentre difetti nell’ossidazione mitocondriale degli acidi grassi può determinare la seconda condizione e possono essere responsabili per le comuni forme di insulino-resistenza. Fig.1 Probabile meccanismo mediante il quale la disfunzione mitocondriale induce insulino-resistenza nel muscolo scheletrico. Una diminuzione dell’ossidazione mitocondriale degli acidi grassi dovuta ad una disfunzione mitocondriale e/o ad una riduzione del numero dei mitocondri, produce un aumento dei livelli intracellulari di acilCoA e diacilglicerolo. Queste molecole attivano una proteina chinasi C, la quale attiva una cascata di fosforilazioni su residui di serina compresi quelli del substrato del recettore dell’insulina IRS-1. L’aumentata fosforilazione su residui critici di serina (es. il residuo Ser 307) blocca la fosforilazione di IRS-1 sui residui di tirosina operata dal recettore dell’insulina (che in seguito al legame con l’insulina acquisisce attività tirosina-chinasica) e di conseguenza inibisce l’attività della fosfatidil-inositolo 3 chinasi. Questa inibizione determina la soppressione del trasporto del glucosio in risposta all’insulina. DISFUNZIONI MITOCONDRIALI, ACIDI GRASSI INTRACELLULARI E INSULINO-RESISTENZA E’ noto che la funzione mitocondriale è richiesta per la normale secrezione dell’insulina stimolata dal glucosio da parte delle cellule β. Recenti studi suggeriscono che molti lievi difetti nella funzione mitocondriale possono avere un ruolo nella patogenesi dell’insulino-resistenza e nel diabete di tipo 2. Petersen et al hanno trovato che, rispetto ai controlli giovani, i soggetti anziani magri e sani hanno una severa insulino-resistenza nel tessuto muscolare, così come un livello significativamente più alto di trigliceridi sia nel muscolo sia nel fegato. Questi cambiamenti sono accompagnati da una diminuzione sia dell’attività ossidativa dei mitocondri sia della sintesi di ATP mitocondriale. Questi dati supportano l’ipotesi che l’insulino-resistenza nell’uomo derivi da difetti nell’ossidazione mitocondriale degli acidi grassi che, di conseguenza, porta ad un aumento di metaboliti degli acidi grassi intracellulari (acilCoA e diacilglicerolo) che alterano il segnale dell’insulina. Alterazioni nel DNA mitocondriale sono state correlate con l’invecchiamento e un recente studio su topi manipolati geneticamente ha fornito evidenze che tali alterazioni possono giocare un ruolo nell’invecchiamento. Se le disfunzioni mitocondriali viste negli anziani studiati da Petersen siano o meno legate alle mutazioni del DNA mitocondriale accumulate negli anni non è ancora chiaro. Altri studi usando la tecnica MRS hanno rilevato simili diminuzioni nell’attività mitocondriale e aumenti del contenuto di grasso intramiocellulare in giovani con insulino-resistenza figli di genitori con diabete di tipo 2, i quali hanno una forte tendenza a sviluppare diabete più tardi nella vita. Inoltre, rispetto ai controlli insulino-sensibili, i soggetti insulino-resistenti hanno una più bassa percentuale di fibre di tipo 1 rispetto alle fibre di tipo 2. Le fibre di tipo 1 sono principalmente ossidative e contengono più mitocondri rispetto a quelle di tipo 2 che sono più glicolitiche. Questi individui possono avere meno mitocondri nei muscoli, probabilmente perché c’è una diminuzione di espressione di geni nucleari che regolano la biogenesi mitocondriale quali il coattivatore 1α e 1ß del perossisoma proliferator-activated receptors (PGC 1 α e PGC 1β). Studi di microarray supportano questa idea: i geni responsivi a PGC 1 α e PGC 1β sono essi stessi down-regolati nei Caucasici obesi con un’alterata tolleranza al glucosio e diabete di tipo 2, e PGC 1 α e PGC 1β sono essi stessi down-regolati sia negli obesi diabetici sia nei messicaniamericani sovrappeso non diabetici. In alternativa la riduzione dell’attività di fosforilazione ossidativa mitocondriale negli individui insulino-resistenti può essere dovuta, non ad una diminuzione del numero dei mitocondri, ma piuttosto ad un difetto della funzione mitocondriale. Questa ipotesi è supportata da studi su biopsie muscolari. In uno studio, l’attività degli enzimi ossidativi mitocondriali era più bassa nei soggetti affetti da diabete di tipo 2 mentre in un altro l’attività della NADH ossidoreduttasi mitocondriale era più bassa. Comunque, a differenza degli studi di MRS, questi studi erano condotti sui mitocondri isolati ottenuti da soggetti diabetici che erano anche obesi. Dal momento che individui obesi hanno mitocondri più piccoli con una ridotta capacità bioenergetica rispetto a quelli dei controlli magri, le anomalie mitocondriali in questi soggetti potrebbero essere in relazione allo stato di obesità piuttosto che all’insulino-resistenza. Il ruolo dell’obesità nella downregolation di PGC 1 α e PGC 1β discussa sopra,è una domanda importante a cui bisogna ancora dare una risposta. Fig 2 Probabile meccanismo mediante il quale la disfunzione mitocondriale mediata da UCP-2 compromette la secrezione di insulina da parte delle cellule beta pancreatiche. A: la proteina UCP2, l’anione superossido e la secrezione di insulina stimolata dal glucosio. La secrezione di insulina è accoppiata al metabolismo del glucosio dall’aumento del rapporto ATP/ADP in seguito all’ossidazione del glucosio, che determina la chiusura del canale del potassio (K). In questo modo la membrana si depolarizza, si apre un canale del Ca++, il successivo ingresso di calcio determina la secrezione dell’insulina. Ucp-2 fa diminuire la secrezione di insulina indotta dal glucosio perché disaccoppia l’ossidazione del glucosio dalla fosforilazione ossidativa e quindi si produce meno ATP. Il superossido generato dall’attività della catena di trasporto degli elettroni stimola l’attività di UCP-2. B: Gli effetti dell’obesità, dell’iperglicemia e dei lipidi si UCP-2. L’ obesità, iperglicemia e gli alti livelli di lipidi inducono ognuno l’espressione di UCP-2 nelle cellule pancreatiche. Questi stimoli fanno aumentare anche la produzione di superossido da parte della catena di trasporto. Come conseguenza UCP-2 viene attivata portando ad un aumento della fuga di protoni mediata da UCP-2. Questa fuga di protoni altera la secrezione di insulina in risposta al glucosio determinando quindi disfunzioni della cellula beta. DISFUNZIONI MITOCONDRIALI E SECREZIONE DELL’INSULINA NELLE CELLULE β PANCREATICHE Molti individui obesi con marcata insulino-resistenza non sviluppano diabete. In questi individui le cellule β-pancreatiche si adattano a rispondere alla più elevata domanda di insulina. Questo adattamento coinvolge l’espansione della massa cellulare β, così come il mantenimento di una normale responsività delle cellule beta al glucosio. In contrasto, negli individui obesi destinati a sviluppare il diabete di tipo 2, le cellule β non secernono abbastanza insulina per compensare l’aumentata richiesta. Questo difetto delle cellule èβ probabilmente causato da una inadeguata espansione della massa cellulare β e/o da un difetto della massa cellulare β esistente a rispondere al glucosio. La massa cellulare β è regolata da vari fattori tra cui le dimensioni delle cellule β, la velocit à di replicazione e/o differenziamento delle cellule β, la velocità della morte cellulare per apoptosi delle celluleβ. Nonostante le difficoltà a quantificarla, la massa cellulare β sembra diminuita negli individui con diabete di tipo 2 rispetto a quella osservata in individui con gradi simili di insulinoresistenza. Benchè le cause di questa diminuzione relativa nella massa delle cellule β sia sconosciuta, l’aumentata velocità di apoptosi può avere un ruolo importante. I segnali da e per i mitocondri che regolano l’apoptosi nelle cellule β e l’effetto del mezzo prediabetico su questi segnali sono completamente sconosciuti, ma saranno probabilmente oggetto di studi futuri. Numerosi studi hanno documentato che in individui con diabete di tipo 2 le cellule beta non percepiscono propriamente il glucosio e quindi non rilasciano appropriate quantità di insulina. La percezione del glucosio richiede il metabolismo ossidativo mitocondriale che porta alla generazione di ATP. Questo aumento del rapporto ATP/ADP nelle cellule beta porta ad una serie di eventi: inibizione del canale del potassio regolato dal rapporto ATP/ADP, la depolarizzazione di membrana, l’apertura di un canale del calcio, afflusso di calcio e la secrezione di insulina. Anche se la secrezione di insulina è modulata anche da altri stimoli che operano indipendentemente, è chiaro che il metabolismo ossidativo mitocondriale ha un ruolo centrale nella secrezione dell’insulina stimolata dal glucosio. Il ruolo critico dei mitocondri è evidente nei rari disordini mitocondriali ereditari in cui le disfunzioni delle cellule beta sono state ricondotte a specifiche mutazioni del genoma mitocondriale. Dato il ruolo centrale del mitocondrio nel percepire il glucosio, è probabile che una diminuzione della funzione mitocondriale nelle cellule beta, analogamente a quanto osservato nel muscolo scheletrico, potrebbe predisporre gli individui a sviluppare disfunzioni delle cellule beta e diabete di tipo 2. Comunque questa interessante ipotesi non è ancora stata verificata sperimentalmente dal momento che è difficile avere campioni di cellule beta. La disfunzione delle cellule beta si pensa sia secondaria ad una aumentata esposizione delle cellule beta al glucosio (glucotossicità) e/o ai lipidi (lipotossicità), frequentemente associata con l’obesità e lo stato di insulino-resistenza. Un certo numero di ipotesi sono state proposte per spiegare come queste condizioni inducano disfunzioni alle cellule beta. Una di queste ipotesi si focalizza sui cambiamenti nell’espressione e nella funzione della proteina della membrana mitocondriale interna UCP-2. Per capire il ruolo di UCP-2 è necessario riprendere alcuni aspetti di rilievo del metabolismo ossidativo. Il metabolismo ossidativo del glucosio coinvolge il trasferimento dell’energia contenuta all’interno dei legami del carbonio del glucosio al terzo legame fosfato dell’ATP. Questa reazione complessa inizia con il trasferimento degli elettroni dei legami carbonio al NAD e al FAD i quali poi li donano alla catena di trasporto degli elettroni, una unità multiproteica divisa in 4 complessi (I, II, III, IV), tutti localizzati nella membrana mitocondriale interna. Infine gli elettroni sono condotti alla loro missione finale, la riduzione dell’ossigeno ad acqua. I complessi I, III e IV sono pompe protoniche guidate da ossidoriduzioni che usano l’energia portata dagli elettroni per pompare protoni fuori dalla matrice, creando un gradiente di potenziale elettrochimico protonico attraverso la membrana interna. Questi protoni poi rientrano nella matrice mitocondriale via ATP-sintasi con l’uso dell’energia immagazzinata all’interno del gradiente elettrochimico per guidare la sintesi di ATP da ADP. L’UCP-2 è una proteina integrale di membrana che, quando attivata, trasporta protoni attraverso la membrana interna, disaccoppiando il metabolismo ossidativo del glucosio dalla produzione di ATP. Dal momento che questo fa diminuire la quantità di ATP generata dal glucosio, l’UCP-2 può regolare negativamente la secrezione di insulina stimolata dal glucosio. Evidenze sperimentali hanno mostrato che ciò è possibile. La forzata iperespressione di UCP-2 nelle cellule beta in coltura determina una diminuzione della secrezione di insulina in risposta al glucosio, mentre l’inattivazione del gene UCP-2 nel topo ha l’effetto contrario. Da notare che l’inattivazione di un solo allele (eterozigosità) produce un effetto che è intermedio tra quello osservato nel topo wild type e nell’omozigote, indicando che cambiamenti relativamente piccoli nell’espressione di UCP-2 hanno effetti significativi nella secrezione di insulina in risposta al glucosio. UCP-2 esercita sostanziali effetti negativi nel controllo della secrezione di insulina in risposta al glucosio. Può la aumentata espressione di UCP-2 avere un ruolo causale nelle disfunzioni delle cellule beta nel diabete di tipo 2? Questa idea è supportata dal fatto che l’espressione di UCP-2 è stimolata in vivo ed in vitro dall’iperglicemia e dai lipidi, oltre che in modelli animali di diabete di tipo 2. La deficienza genetica di UCP-2 nei modelli animali di obesità/diabete determina un significativo miglioramento della funzione delle cellule beta. Questi dati suggeriscono che UCP-2 nei modelli animali ha un importante ruolo patogenetico. Un ruolo simile potrebbe averlo anche nel diabete di tipo 2 umano, perché UCP-2 è espresso dalle cellule beta e la sua espressione aumenta con l’iperglicemia. Inoltre un polimorfismo nel promotore del gene UCP-2 umano, che incrementa l’espressione di UCP-2, è stato legato alla riduzione della secrezione di insulina e ad una più alta frequenza e/o più precoce comparsa del diabete di tipo 2. E’ stato recentemente scoperto che il superossido, un sottoprodotto dell’attività della catena di trasporto degli elettroni, stimola l’attività della UCP-2 quando viene aggiunto a mitocondri isolati, o quando è generato in situ all’interno delle cellule beta intatte. Il meccanismo attraverso il quale il superossido attiva UCP2 è sconosciuto ma può coinvolgere la generazione di intermedi dei radicali liberi. La stimolazione dell’attività di UCP-2 da superossido è rilevante nello sviluppo delle disfunzioni delle cellule beta, perché la produzione di superossido aumenta nelle cellule beta dei roditori con diabete di tipo 2 ed in cellule beta in coltura esposte all’iperglicemia e a elevati livelli di lipidi. Questo aumento di perossido, accoppiato all’aumento nella proteina UCP-2, porta ad una marcata stimolazione della fuga di protoni che porta alla disfunzione delle cellule beta. La rimozione del superossido endogeno nelle cellule beta che non rispondono al glucosio, sia perché esposte all’iperglicemia in vitro o perché in presenza di uno stato diabetico o di obesità in vivo, inibisce notevolmente l’attività di UCP-2 e ripristina la secrezione di insulina stimolata dal glucosio. Queste evidenze suggeriscono la possibilità che inibitori di UCP-2 possano essere usati per trattare o prevenire il diabete di tipo 2. CONCLUSIONI Molti diversi esperimenti supportano l’ipotesi che i difetti mitocondriali possano giocare un ruolo critico nei due principali aspetti del diabete di tipo 2. L’insulinoresistenza e la disfunzione delle cellule beta. Varie domande restano ancora aperte: 1) la riduzione in vivo della funzionalità mitocondriale è dovuta alla perdita di mitocondri, o a difetti funzionali dei mitocondri o a entrambe le cose? 2) La down-regolazione dei geni responsivi a PGC-1α/PGC-1β è un evento primario o secondario nella patogenesi del diabete di tipo 2? Se è un evento primario, quali sono i geni a monte responsabili della loro alterata espressione? 3) Può l’UCP-2 giocare un ruolo nella disfunzione della cellula β pancreatica nei pazienti con diabete di tipo 2? Rispondere a queste domande potrà fornire nuovi bersagli farmacologici per la prevenzione e il trattamento della più diffusa malattia metabolica del mondo.
Scarica