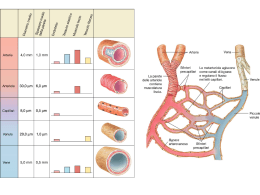
Anomalie Vascolari di interesse pediatrico Carlo Gelmetti*, Riccardo Cavalli** Dipartimento di Fisiopatologia Medico‑Chirurgica e dei Trapianti – Università degli Studi di Milano *; Fondazione IRCCS Ca’ Granda “Ospedale Maggiore Policlinico”*,** Con il termine angiomi vengono indicate impropriamente quasi tutte le anomalie vascolari di interesse dermatologico che sono attualmente divise in due categorie principali: le malformazioni vascolari e i tumori vascolari. Qui verranno trattate solo le anomalie vascolari di interesse pediatrico più comuni. La nuova classificazione adottata dall’ISSVA (International Society for the Study of Vascular Anomalies) nel congresso di Melbourne del 2014 costituisce un’evoluzione di quella, più nota, del 1996 (Tab. 1). In essa, i tumori vascolari vengono suddivisi in 3 gruppi in relazione al grado di aggressività neoplastica. Le malformazioni vascolari sono distinte in semplici e combinate, elencando queste ultime in maniera dettagliata. Vengono inoltre inserite anche le malformazioni dei vasi maggiori. La causa delle anomalie vascolari non è nota, ma non è imputabile né a traumi da parto, dato che esse si manifestano anche nei cesarizzati, né, tantomeno, a particolari desideri delle gravide (da cui il termine popolare di “voglie”). Le malformazioni vascolari (MV) sono anomalie strutturali, errori congeniti della morfogenesi vascolare, che avvengono nel corso dell’embriogenesi e mostrano un turnover cellulare endoteliale normale. La sindrome di Sturge‑Weber è causata da una mutazione post‑zigotica non ereditaria del gene GNAC. Anomalie del gene RASA 1 sono state descritte in alcune forme di teleangiectsie e malformazioni artero‑venose. Tra i tumori vascolari, gli Emangiomi Infantili (EI) presentano un marcatore chiamato GLUT‑1 (una proteina di trasporto del glucosio), che è assente nelle MV ed è invece presente a livello placentare. La teoria che supporta la relazione tra gli EI e la placenta ipotizza che le cellule di origine placentare possano embolizzare i tessuti fetali ricettivi come la cute nel corso della gestazione o alla nascita. Questa ipotesi potrebbe spiegare l’incidenza di EI dopo un prelievo dei villi coriali e l’associazione frequente degli EI con la prematurità o un basso peso alla nascita, condizioni che sono correlate a danni placentari e che potrebbero causare un distacco e una disseminazione intravascolare di cellule placentari. In alternativa si sospetta l’ipossia fetale come possibile trigger. Nel 2014 è anche (finalmente!) arrivata l’approvazione del propranololo per la cura degli emangiomi infantili sia da parte della FDA che della EMA. MALFORMAZIONI VASCOLARI Le malformazioni vascolari (MV) sono anomalie strutturali, errori congeniti della morfogenesi vascolare che avvengono nel corso dell’embriogenesi e mostrano un turnover endoteliale normale. Sebbene le MV siano per definizione presenti alla nascita, alcune si manifestano nell’adolescenza o nell’età adulta. Esse possono essere suddivise 1 anatomicamente secondo il tipo di vaso interessato in sottogruppi: capillari, venose, arteriose e linfatiche; inoltre, possono associarsi dando origine a forme complesse. È importante anche differenziare le MV a bassa portata, le più frequenti, da quelle ad alta portata, in pratica solo le MV arteriose e le fistole artero‑venose. Malformazioni vascolari capillari. Le MV capillari sono chiamate comunemente angiomi piani o nevi flammei o anche nevi “a vino di porto”. Tranne in casi eccezionali, sono presenti dalla nascita, persistono per tutta la vita e colpiscono i due sessi in uguale misura. La loro frequenza, se non si considerano le MV capillari della linea mediana, è bassa (0,3%). Per la loro natura possono presentare associazioni patologiche. Le MV capillari colpiscono la cute e anche le mucose con chiazze di dimensioni variabili da pochi millimetri fino a coinvolgere più dermatomeri. Il capo è la regione più colpita. Le più comuni MV capillari sono le chiazze vascolari dei neonati chiamate anche nevi flammei o salmon patches, a localizzazione medio‑frontale, palpebrale o nucale, che si attenuano con il tempo e non comportano disturbi estetici importanti. Le chiazze nucali (nevo di Unna, “morso della cicogna”) si riscontrano fino all’80% dei neonati caucasici e sono da considerarsi parafisiologiche, come la chiazza medio‑frontale. Se le chiazze sacrali sono accompagnate da nevi o fistole o noduli, possono essere associate a malformazioni come la spina bifida. Le chiazze vascolari mediane si rendono poco visibili già dopo il primo anno di età. Le MV capillari laterali costituiscono invece un disturbo estetico ingravescente; infatti, il colore con il tempo si scurisce diventando rosso‑violaceo. Cosa ancora più importante, esse possono essere facilmente una spia di anomalie malformative complesse. Il nevo anemico può essere considerato il contrario di una MV capillare; è infatti una malformazione funzionale caratterizzata da una chiazza, di solito unica, di forma irregolare con un bordo dentellato, più pallida della cute circostante. Si distingue dal nevo acromico perché lo sfregamento della cute non induce vasodilatazione. Recentemente è stato associato alla NF1. Angioma stellare. Chiamato anche nevus araneus, spider nevus, l’angioma stellare più che una malformazione viene considerata una dilatazione di un vaso preesistente ed è una lesione comunissima, soprattutto nei bambini e nei giovani. Si presenta come un punto rosso di circa 1 mm circondato da un alone eritematoso, che è costituito da fini teleangectasie disposte a raggi come una piccola stella o un piccolo ragno, da cui i due nomi. Una diascopia leggera fa scomparire l’alone e indica che il punto centrale è una piccola arteria, perché è pulsatile; una diascopia più energica fa scomparire anche il punto centrale. Non si conosce la causa di queste lesioni, ma il fatto che siano quasi sempre localizzate su cute esposta fa pensare che siano il risultato di microtraumi. In alcuni casi gli angiomi stellari scompaiono da soli. In genere costituiscono solo un problema estetico e possono comunque essere eliminati con una lieve elettrofolgorazione o con laser. Teleangectasie. Sono MV capillari asintomatiche in forma di piccole ectasie lineari, arboriformi o in chiazzette puntiformi simulanti un angioma stellare. Sono da considerarsi 2 parafisiologiche negli anziani quelle del volto e del collo, indotte essenzialmente dal fotoinvecchiamento, e quelle degli arti inferiori (teleangectasie arboriformi), indotte dalla pressione idrostatica e riscontrabili soprattutto nelle donne. La teleangectasia nevoide unilaterale e, soprattutto, la teleangectasia essenziale generalizzata vanno distinte dalla malattia di Rendu‑Osler, o teleangectasia emorragica ereditaria. In quest’ultima, le lesioni si sviluppano più lentamente e, inoltre, la cute, le mucose e gli organi interni presentano frequenti sanguinamenti. L’atassia‑teleangectasia (sindrome di Louis‑Bar) è una malattia a trasmissione ereditaria autosomica recessiva caratterizzata da atassia, teleangectasie cutanee e oculari e frequenti infezioni respiratorie. I pazienti hanno diminuiti tassi di immunoglobuline, anomalie strutturali del timo e dei linfonodi e aumentato rischio di linfoma e di leucemia a cellule T. Cutis Marmorata Teleangectasica Congenita (CMTC). Detta anche sindrome di Van Lohuizen, costituisce una rara anomalia capillaro‑venosa del derma e del tessuto sottocutaneo. Le lesioni colpiscono più spesso gli arti con striature violacee serpiginose a disposizione reticolare, che si accentuano con il freddo e la posizione declive; regrediscono parzialmente nei primi mesi di vita, lasciando atrofie più o meno accentuate. Sono state segnalate numerose anomalie associate. La CMTC va distinta dalla livedo fisiologica del neonato, che però è omogeneamente diffusa, si presenta in seguito a un abbassamento della temperatura per poi regredire in pochi minuti, se le condizioni ambientali si normalizzano, e si manifesta con un reticolo regolare a piccole maglie. La sede relativamente profonda della lesione rende quasi inutile il trattamento della CMTC con il laser vascolare. Angiocheratomi. Sono MV congenite dovute a ectasie dei capillari subepidermici con una reazione epidermica proliferativa. Gli angiocheratomi sono papule ipercheratosiche rosse, violacee o nere del diametro di 1‑10 mm. Sono stati descritti vari tipi di angiocheratomi. L’angiocheratoma circoscritto costituisce la sola forma che può essere presente alla nascita ed è più frequente nel sesso femminile. Le lesioni papulose o nodulari sono unilaterali e localizzate agli arti inferiori o al tronco, raramente agli arti superiori. L’angiocheratoma di Mibelli è una malattia autosomica dominante che si manifesta nell’adolescenza e prevalentemente nel sesso femminile, sulle mani e sui piedi, ma può anche colpire i gomiti, le ginocchia, le caviglie e le natiche. Sono spesso associati acrocianosi e geloni. L’Angiocheratoma corporis diffusum è una rara forma generalizzata che può essere idiopatica o associata a varie patologie metaboliche. La più nota è la malattia di Fabry, trasmessa con eredità recessiva legata al sesso, dovuta a un disordine degli sfingolipidi (deficienza di a galattosidasi A), che provoca un accumulo di ceramide‑triexoside in cellule endoteliali, fibroblasti, periciti, cellule muscolari dei vasi sanguigni del cuore, neuroni e cellule renali. Si manifesta nell’infanzia o nella prima adolescenza, con lesioni che vanno da piccole macchie teleangectasiche a papule verrucose, disseminate soprattutto su tronco e scroto e alla radice degli arti. I pazienti presentano il fenomeno di Raynaud, dolori articolari, ipertensione, proteinuria, cardiomegalia con elevato rischio di mortalità. Malformazioni vascolari venose. Le MV venose comprendono lesioni congenite o ad apparizione tardiva e possono essere isolate, regionali o diffuse. Costituiscono un’ampia 3 gamma di anomalie che va dalle varicosità o flebectasie cutanee a masse cutanee spugnose e a lesioni complesse che coinvolgono più tessuti. In superficie sono visibili come masse elastiche violacee, più o meno lobulate, indolori e svuotabili alla pressione. In profondità si palpano delle masse elastiche indolenti e la pelle soprastante può essere normale o assumere una sfumatura bluastra. Fleboliti possono essere palpati e osservati radiologicamente. Le MV venose possono essere compresse e diminuire di dimensione; aumentano invece di dimensione in posizione declive o con la manovra di Valsalva. Malformazioni vascolari artero‑venose. Sono più spesso masse profonde di varia dimensione ricoperte da cute normale o da uno pseudoangioma piano, oppure sembrano normali angiomi che però non regrediscono come dovrebbero nei primi ann. Si tratta di malformazioni ad alta portata che a volte compaiono nel periodo neonatale, più spesso dopo qualche anno di vita. Una MV artero‑venosa si sviluppa gradualmente nell’infanzia, ma può peggiorare all’improvviso in seguito a traumi locali, ad asportazione chirurgica, a legature vascolari e a stimoli vari (pubertà, gravidanza, farmaci). La cute frequentemente è ipertermica, presenta chiazze di capillari dermici soprastanti, può ulcerarsi con emorragia e i pazienti possono avvertire dolori pulsanti. All’esame obiettivo si percepisce un fremito al tatto e un soffio all’ascoltazione. Malformazioni vascolari linfatiche. Le MV linfatiche in genere sono lesioni papulo‑ vescicolari translucide (perlomeno quando sono localizzate vicino alla superficie cutanea), che tendono a confluire. I due terzi sono già evidenti alla nascita e il 90% compare prima dei 2 anni di età. Le lesioni assumono varie forme. Alcune sono puramente cistiche (igromi cistici, cisti linfatiche) e sono più frequenti al collo; altre sono formate da tessuto linfatico compatto e sono predominanti alla faccia e alla lingua; altre, infine, sono formate da un’associazione dei due tipi con micro‑ e macrocisti immerse in tessuto compatto. Altre sedi interessate in ordine di frequenza sono le regioni ascellare e mediastinica, ma anche alcuni organi interni. Nelle MV linfatiche si possono associare ipertrofia e deformazione scheletrica. Alcune sembrano essere evolutive sia per le recidive dopo la terapia, sia per episodi infettivi o infiammatori. Malformazioni vascolari complesse. Le malformazioni vascolari a bassa portata (capillari, venose e linfatiche) sono frequentemente associate a una crescita ossea esagerata nella regione cranio‑facciale e alle estremità. Queste anomalie sono talvolta accompagnate da chiazze brune (maculose o verrucose), da lipomi, ipertricosi, iperidrosi, displasie valvolari venose e malformazioni spongiose multiple dette flebangiomi, che possono coinvolgere i visceri. La terapia è individuale e quasi sempre multidisciplinare. Terapia. La cura delle MV è sostanzialmente chirurgica e include laserterapia esterna o intraluminale, embolizzazione o chirurgia vera e propria. La elettrocoagulazione rimane un’opzione, anche se limitata dalla disponibilità di un laser vascolare La terapia delle MV capillari mira a distruggere i capillari ectasici senza danneggiare le strutture circostanti; i laser vascolari (dye laser a 590 nm; Nd:YAG a 532 nm) danno buoni risultati nella maggior parte dei pazienti, con una discreta tollerabilità locale e una quasi totale assenza di effetti collaterali come cicatrici e discromie residue. Di recente introduzione e anche la metodica sequenziale che prevede l’erogazione in rapida sequenza di un doppio impulso Dye‑ 4 Nd:YAG: i due impulsi a diversa lunghezza d’onda (rispettivamente 595‑1064 nm) emessi a distanza di una frazione di secondo consentono la preliminare trasformazione dell’ossiemoglobina in metaemoglobina e una successiva penetrazione della radiazione Nd:YAG fino ad una profondità di 7‑8 mm. La metodica sequenziale e indicata per il trattamento delle malformazioni capillari ipertrofiche o resistenti al trattamento con Dye laser ed e efficace anche per il trattamento di altre anomali vascolari come ad esempio angiocheratomi, granulomi piogenici e spider nevi.Il trattamento laser può essere intrapreso in età pediatrica e addirittura in epoca neonatale. Un’anestesia di superficie con lidocaina e prilocaina riduce la sensazione di dolore, ma non sempre è sufficiente. I laser vascolari più moderni dispongono di sistemi di raffreddamento del raggio laser. Quando è però necessaria l’immobilità assoluta (per esempio, con lesioni orbitarie), bisogna anestetizzare il bambino. La terapia delle MV arteriose, venose e linfatiche è di competenza del chirurgo vascolare, anche se recentemente sono stati trattati con successo dei casi di linfangiomi con sildenafil orale. TUMORI VASCOLARI Emangiomi infantili (EI). Costituiscono un gruppo ben definito di tumori vascolari benigni che, proliferando nei primi mesi di vita, costituiscono una massa cellulare che ha bisogno di canali vascolari neoformati per essere alimentata e drenata (fase di proliferazione). Questi vasi possono essere predominanti in alcuni emangiomi cutanei o epatici voluminosi, simulando una malformazione vascolare ad alta portata. Tutti gli emangiomi capillari subiscono una regressione spontanea nell’arco di qualche anno (fase d’involuzione). Gli EI colpiscono circa il 10% dei lattanti, soprattutto i prematuri di basso peso, e sono nettamente più frequenti nel sesso femminile (F: 3/1; M: 5/1). La maggior parte degli EI viene notata dopo i primi giorni di vita come una chiazza eritematosa, oppure come una chiazza ischemica (herald patch) a tipo nevo anemico, o ancora come teleangectasie circondate da un alone ischemico. La maggioranza degli EI è costituita da una singola lesione, di solito cutanea, anche se esistono angiomi viscerali, in genere epatici. Dal punto di vista topografico, il capo è la regione più colpita. Le dimensioni variano da lesioni puntiformi a forme estese, segmentarie, che interessano ampie regioni. Dopo l’esordio, gli EI si sviluppano rapidamente in diametro e in volume fino a 4‑6 mesi di vita, raramente fino a 9‑12 mesi. Negli ultimi anni si è sottolineata l’importanza della morfologia e della topografia degli EI, in particolare per quanto riguarda il livello di rischio degli EI stessi. Secondo questo modo classificativo, gli EI possono essere descritti in due forme: localizzati e segmentari. I primi, la stragrande maggioranza, sono tipicamente nodulari e rotondeggianti e non hanno associazioni patologiche, mentre i secondi (EI segmentari) si manifestano come placche rilevate a distribuzione simil‑dermatomerica e possono associarsi con altre patologie (come le sindromi PHACES oppure PELVIS/SACRAL/LUMBAR)1 oltre ad andare più facilmente incontro a ulcerazioni. Va 1 PHACES (Posterior fossa malformations, Hemangioma, Arterial anomalies, Cardiac abnormalities/aortic coartation, Eye abnormalities, Sternal cleft defects) e la sindrome PELVIS (Perineal hemangioma, External genitalia malformations, Lipomyelomeningocele, Vesicorenal abnormalities, Imperforate anus, Skin tag); 5 aggiunto che esistono anche gli EI multifocali (quando sono presenti più di 6 lesioni angiomatose) in cui vanno depistate eventuali lesioni angiomatose viscerali (fegato). Nella fase di stato, gli EI assumono tre aspetti clinici: superficiale, sottocutaneo (profondo) e misto. In fase di stato, la forma superficiale (il cosiddetto angioma tuberoso o angioma a fragola), si presenta come una tumefazione cupoliforme di colore rosso vivo o rosso vinoso, più o meno lobulata, calda al tatto, di consistenza elastica o duro‑elastica, che aumenta leggermente di volume e consistenza sotto sforzo o durante il pianto e che non si svuota alla pressione. Dopo qualche mese di apparente stabilità, la lesione comincia a regredire al centro con aree più chiare, grigiastre, che progrediscono in senso centrifugo, dando un aspetto più pallido alla lesione, che contemporaneamente diminuisce di consistenza, comincia ad afflosciarsi e diventa meno calda. La regressione spontanea si completa di norma verso i 5‑7 anni. L’EI sottocutaneo o profondo, chiamato impropriamente cavernoso, è visibile come una massa elastica ben delimitata, poco depressibile e calda, ricoperta da epidermide normale o con tenue sfumatura cianotica. Esso pone problemi di diagnosi differenziale con una malformazione venosa o un linfangioma cistico o un encefalocele. Nella maggior parte dei casi l’EI profondo si associa a quello superficiale, facilitando la diagnosi. Si ricorda che la sede profonda, in assenza di un angioma sentinella superficiale, ritarda molto l’osservazione della lesione che non viene notata, di solito, se non dopo i primi mesi di vita. La regressione spontanea degli EI lascia esiti minimi nelle lesioni più piccole, mentre, in quelle maggiori, può dare luogo a sequele di varia entità (cicatrici, teleangectasie, chiazze ipoelastiche giallastre ecc.), soprattutto all’estremo cefalico, anche se è difficile prevedere la qualità del risultato finale. Oltre all’alterazione estetica, l’EI può ulcerarsi e formare un’escara con esito cicatriziale. Nelle regioni perioculari si possono avere disturbi della vista (per esempio, ambliopia). Un EI a localizzazione laringea può dare polmonite ab ingestis e soffocamento. L’angioma Cyrano è un EI che colpisce la punta del naso rendendola rossa e rigonfia, da cui il nome. L’importanza della sede consiglia la pronta terapia medica per limitare al minimo l’espansione della lesione e la diastasi delle cartilagini nasali. In caso di esiti, la chirurgia plastica va eseguita preferibilmente nel corso del terzo anno di vita. L’angioma cutaneo‑ laringeo è un EI che coinvolge bilateralmente il territorio della terza branca trigeminale “a barba”, e si estende all’interno della cavità orale fino alla laringe, con possibilità di ostruzione delle vie aeree. Il suo sanguinamento può essere pericoloso. Se l’angioma è troppo esteso e/o risponde in modo insufficiente alla terapia medica, bisogna tracheostomizzare il paziente. L’emangiomatosi neonatale diffusa è una rara condizione caratterizzata dalla presenza, alla nascita o nelle settimane successive, di decine o centinaia di EI su tutto l’ambito cutaneo e negli organi interni, che può comportare diverse complicanze come emorragie intestinali, scompenso cardiaco, compromissione delle vie aeree e del sistema nervoso. Esiste anche una forma benigna (emangiomatosi benigna neonatale), con soli elementi cutanei ed epatici o anche solo cutanei. Sebbene, data la loro SACRAL (Spinal Dysraphism, Anogenital, Cutaneous, Renal and Urologic Anomalies, Associated with an Angioma of Lumbosacral Localization); LUMBAR (Lower body hemangioma/ Lipoma or other cutaneous anomalies, Urogenital anomalies, Myelopathy, Bony deformities, Anorectal/Arterial anomalies, and Renal anomalies) 6 natura, gli EI non si associno quasi mai a malformazioni, la sindrome PHACES e la sindrome PELVIS altrimenti detta SACRAL o LUMBAR sono le eccezioni che confermano la regola. Emangiomi Congeniti. A differenza degli EI che insorgono quasi sempre nella prima‑ seconda settimana di vita, gli emangiomi congeniti sono completamente sviluppati già alla nascita e si presentano come masse rosso‑violacee, rotondeggianti od ovalari, calde ed elastiche. In base al loro comportamento, si dividono in due gruppi: RICH (Rapidly Involuting Congenital Hemangioma) e NICH (Non Involuting Congenital Hemangioma). Le lesioni sono istologicamente entrambe GLUT‑1 negative e non hanno preferenza di sesso a differenza degli EI che sono in gran parte a carico delle femmine, ma, mentre i RICH si afflosciano entro il primo anno di vita (più rapidamente degli EI classici), i NICH persistono invariati. Granuloma piogenico. Detto anche granuloma teleangectasico, è una neoformazione costituita da un piccolo nodulo rosso tondeggiante o finemente lobulato a superficie liscia che insorge repentinamente, quasi sempre in sedi esposte come volto, collo e arti. Può insorgere a qualsiasi età, ma è più comune nei bambini. La crescita rapida, erode l’epitelio, per cui al minimo trauma segue un sanguinamento apparentemente esagerato rispetto alle dimensioni. Il fatto che sia prevalente in sedi esposte fa ipotizzare una causa di tipo microbiologico. Il trattamento consiste in elettrocoagulazione o laserterapia; si sconsigliano crioterapia e asportazione chirurgica che esitano più facilmente in una satellitosi reattiva. Tumore glomico. È una lesione nodulare con tonalità bluastra di pochi millimetri, incassata, poco compressibile e dolente alla compressione. Dal punto di vista clinico, più spesso si osserva un tumore solitario, in genere localizzato agli arti (tipica la sede sottoungueale), oppure, più raramente, un tumore multiplo (chiamato anche glomangiomatosi), che ha un ereditarietà autosomica dominante. In questo caso i glomangiomi non hanno sede preferenziale e sono meno dolorosi della forma solitaria. Se indicata, la terapia può essere chirurgica. Fenomeno di Kasabach‑Merritt. Si tratta di una rara evenienza dove una neoplasia vascolare (un tufted angioma o un emoangioendotelioma kaposiforme), presente fin dalla nascita o dalle prime settimane di vita, si associa a trombocitopenia, anemia e splenomegalia. Dal punto di vista clinico, quando non è troppo profonda, si osserva una massa di colorito rosso scuro o rosso vinoso, calda, ricoperta da cute tesa e lucida, con possibili discromie periferiche dovute a eventuali fenomeni emorragici causati dal sequestro e dall’aumentata distruzione delle piastrine nelle lacune della lesione. È una patologia grave e, infatti, nonostante l’impiego di varie terapie, alcuni pazienti non rispondono ed evolvono fino all’exitus. TERAPIA L’involuzione spontanea degli EI permette di scegliere l’astensione terapeutica nei casi di lesioni di piccole dimensioni in sedi non orifiziali e non deturpanti. È comunque indicato rassicurare i genitori sul decorso favorevole con l’ausilio di illustrazioni e fotografie che 7 mostrano l’andamento di tali lesioni. Il propranololo (Tab. 2) per via sistemica costituisce la terapia d’elezione nelle seguenti situazioni: • rapido accrescimento della lesione; • emorragie ricorrenti, ulcerazioni o infezioni; • interferenza della lesione con importanti funzioni fisiologiche (respirazione, alimentazione, vista, udito); • presenza di EI di grandi dimensioni o multicentrici in grado di determinare scompenso cardiaco. La terapia con propranololo alla dose di 2‑3 mg/kg/die per 6 mesi è oramai lo standard ed è stata approvata da parte della FDA e della EMA. L’efficacia e la sicurezza di impiego del propranololo sono maggiori rispetto ai corticosteroidi e quindi possono permettere un trattamento degli EI anche non complicati. Nel caso dei corticosteroidi, la terapia orale “ad impulsi” dà i migliori risultati e deve essere fatta nella fase proliferativa dell’EI (mentre il propranololo sembra attivo anche in fase di stato). Nel passato, a riguardo degli EI resistenti e dopo il primo anno di vita, si era considerato l’IF per via sottocutanea (3.000.000 U/m2/ die) per 3‑6 mesi. Tale trattamento rimane ora una possibilità solo nel fenomeno di Kasabach‑Merritt, anche se, in questo caso, è stata segnalata l’efficacia del sirolimus. Analogamente, nei casi di angiomi gravi e resistenti si può impiegare la vincristina . Attualmente, i risultati ottenuti con il propranololo sembrano relegare le alternative terapeutiche a nicchie limitatissime. Utile la laserterapia per accelerare la riepitelizzazione degli EI ulcerati. Per la correzione di esiti antiestetici si consideri la chirurgia plastica. Interessanti appaiono i risultati con altri beta‑bloccanti anche per via topica (es. timololo). Bibliografia 1. George A, Mani V, Noufal A. Update on the classification of hemangioma. J Oral Maxillofac Pathol. 2014;18(Suppl 1):S117‑20. 2. Dasgupta R, Fishman SJ. ISSVA classification. Semin Pediatr Surg. 2014;23(4):158‑61. 3. Wójcicki P, Wójcicka K. Epidemiology, diagnostics and treatment of vascular tumours and malformations. Adv Clin Exp Med. 2014;23(3):475‑84. 4. Kollipara R, Odhav A, Rentas KE et al. Vascular anomalies in pediatric patients: updated classification, imaging, and therapy. Radiol Clin North Am. 2013;51(4):659‑72. 5. Bauman NM, McCarter RJ, Guzzetta PC et al. Propranolol vs prednisolone for symptomatic proliferating infantile hemangiomas: a randomized clinical trial. JAMA Otolaryngol Head Neck Surg. 2014 Apr;140(4):323‑30. 8
Scarica