BIOINFO3 - Lezione 37
1
BIOINFO3 - Lezione 37
2
BIOINFO3 - Lezione 37
3
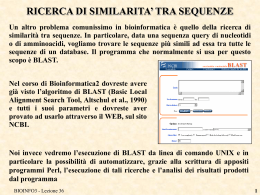
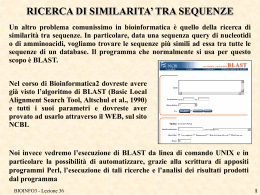
PARSING RISULTATI DI BLAST
Nella lezione di ieri abbiamo visto come automatizzare l’esecuzione di BLAST. Oggi
proviamo ad analizzare automaticamente i risultati prodotti.
Supponiamo quindi in qualche modo di avere a disposizione in una stringa il report
prodotto dall’esecuzione di BLAST. La stringa potrebbe essere stata catturata con il
comando qx di esecuzione del blast
$report=qx{blastall ……};
Oppure se l’output del blastall è stato rediretto su un file (ovviamente anche in un
programma eseguito precedentemente), il file potrebbe essere letto ed il suo contenuto
assegnato alla stringa
qx{blastall …… > file-report}; #anche in un altro programma
…………
open (I,”file-report”);
while ($riga=<I>){
$report.=$riga;
}
BIOINFO3 - Lezione 37
4
REPORT DI BLAST
Proviamo a vedere come è strutturato dal punto di vista sintattico il report prodotto da
BLAST. Bisogna innanzitutto distinguere due casi:
1- Il programma non ha trovato similarità locali significative tra query e database
Versione e
riferimenti
Sequenza query
Database
Pattern da
ricercare
BIOINFO3 - Lezione 37
5
REPORT DI BLAST
2- Sono state trovate delle sequenze del database con similarità locali alla query
Versione e
riferimenti
Sequenza query
Database
Pattern da ricercare
Sequenze con E-value
(probabilità di allineamento
dovuto al caso) minore della
soglia predefinita
Per ciascun allineamento
individuato:
dettaglio e parametri
BIOINFO3 - Lezione 37
6
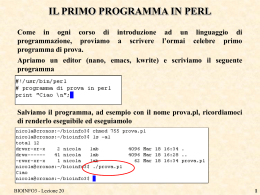
ESERCIZIO
Provate voi a scrivere il programma che legga il file di risultati di
BLAST passato come argomento, cerca il pattern “Sequences
producing...” e per gli allineamenti trovati stampi i valori di:
GI
Length
Score
E-Value
Identities
%Identities
Gaps (se ci sono)
%Gaps (se ci sono)
BIOINFO3 - Lezione 37
7
Ecco un
programma che
individua (se ce ne
sono) le sequenze
(e i parametri
dell’allineamento)
del database che
danno
allineamenti
significativi con la
query
BIOINFO3 - Lezione 37
8
L’array contiene 4 elementi ma il primo verrà scartato
Dopo lo shift l’array contiene i 3 allineamenti
BIOINFO3 - Lezione 37
9
Stampiamo $s,
ovvero tutti i
parametri
dell’allineamento
di una delle
sequenze trovate
La stessa sequenza ha 2
allineamenti con la query! Però,
per semplicità, in questo
programma consideriamo solo il
primo.
BIOINFO3 - Lezione 37
10
Siamo nel primo ciclo
BIOINFO3 - Lezione 37
11
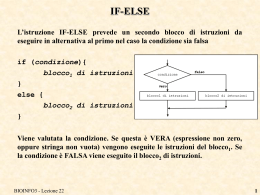
FINE ESECUZIONE
Facciamo continuare (c) il programma fino alla fine e osserviamo come siano
stampati i parametri anche per le altre due sequenze che hanno trovato
allineamento
In questo programma di esempio non facciamo nulla di particolare con i dati
acquisiti, se non stamparli, però potremo ad esempio selezionare le sequenze
in base all’e-value o in base alla percentuale di similarità.
BIOINFO3 - Lezione 37
12
ESTRAZIONE DELLE SEQUENZE
Una volta individuato il codice delle sequenze con similarità alla sequenza
query, potrebbe interessarci, per successive elaborazioni, avere a disposizione
la sequenza completa di tali sequenze.
Un modo per ricavarla, visto che siamo in possesso del codice gi delle
sequenze, potrebbe essere quello di scrivere un programma parserizzatore del
file FASTA originale del database.
Esiste però una possibilità più comoda. Il pacchetto di BLAST offre, tra gli
altri, anche un programma che si chiama fastacmd, che fa già tale lavoro.
fastacmd –d database –s stringa
Il database è quello su cui abbiamo fatto il blast, mentre la stringa fornita è o
il codice gi della sequenza o anche il suo accession. Il programma fastacmd
restituisce in formato FASTA sullo standard output la sequenza del database
individuata da tale codice
BIOINFO3 - Lezione 37
13
FASTACMD
Unica condizione richiesta per poter eseguire il comando fastacmd è quella di
aver formattato il database con l’opzione –o al momento dell’esecuzione del
formatdb di quel database di sequenze.
BIOINFO3 - Lezione 37
14
FASTACMD
Ovviamente
il
comando
fastacmd
potrà essere eseguito
anche da programma,
al pari di tutti gli altri
comandi UNIX.
Vediamo
questo
programma
che
riceve il nome del file
di risultati di BLAST
ed un numero e
stampa (in FASTA)
solo le sequenze con
e-value inferiore o
uguale al numero
fornito
BIOINFO3 - Lezione 37
15
FASTACMD
BIOINFO3 - Lezione 37
16
RIEPILOGO
•Parsing risultati di BLAST
•FASTACMD
BIOINFO3 - Lezione 37
17
Scarica